






 分子动力学计算涉及数千原子规模的系统,计算步骤包括设置初始位置和速度,确保符合玻尔兹曼分布,通过不断迭代和温度校正达到热平衡。适合研究小于1ns时间尺度的反应。初始结构的选择对模拟效果至关重要,通常基于实验数据或能量最小化结果。
分子动力学计算涉及数千原子规模的系统,计算步骤包括设置初始位置和速度,确保符合玻尔兹曼分布,通过不断迭代和温度校正达到热平衡。适合研究小于1ns时间尺度的反应。初始结构的选择对模拟效果至关重要,通常基于实验数据或能量最小化结果。
分子动力学计算流程:
系统容量
在进行分子动力学计算前,必须先估计运 算的可行性,一般分子动力学计算的容量 为数千个原子级别,如果系统过大则超出 计算的范围。
对于N个原子的系统,每计算一步需要计算 1/2N(N+1)组远程作用力,这是运算过程中 最耗时的部分,若原子增加一倍,计算时 间则为1/2×2N(2N+1)为原来的四倍多。
研究对象所处的时间范围:
通常分子动力研究所选取的积分步长为飞秒fs ( 1fs=10-15s),若以目前一般的个人工 作站或较强的个人电脑从事1000个原子系统的计算,累积100万步即研究10-9s ( 1ns)的 时间范围,(需要两星期的时间?)
因此从实际的角度来讲,分子动力学适合研究反应或运动时间小于1ns的体系,而不适合较慢的反应或运动。例如蛋白质折叠 在10-3s ( 1ms)级别 ,则需要非常长的时间。
计算过程
执行分子动力学计算时,将一定数目的分 子放在一定形状的盒子中,并使它的密度 和实验密度相符合,再选定实验的温度, 即可以着手计算。
通常是将分子随机的放置在盒子中,或是 按照其结晶位置排列,这将作为分子动力模拟的初始位置。计算开始时,还必须知道原子的初始速度。系统中所有原子速度产生的动能总和应 当满足玻尔兹曼分布:
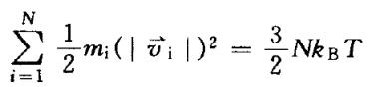
式中N为原子数目, T为热力学温度, kB为 玻尔兹曼常数。原子速度可以按此式随机产生,而总动能 符合3/2NTkB即可。
由初始位置和速度开始,计算每一步新的速度和坐标,再由新的速度计算系统的温度:
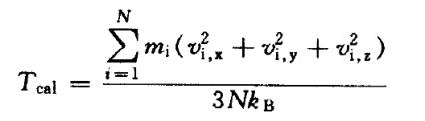
如果得到的速度过大或过小,则会使得到 的温度大于或小于设定的温度多,这时 需要校正速度,一般所允许的温度范围为

如果计算的温度超过允许范围,则将所有
原子的速度乘以一个校正因子:
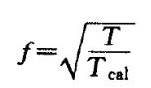
使系统温度重新调整为设定温度:
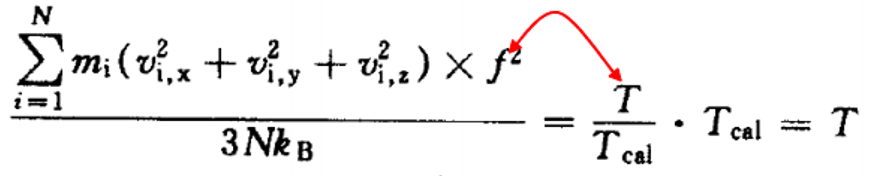
实际的计算中,开始时每隔几步就需要校正一次,随着计 算的进行,校正间隔逐渐增加到几千步,一直到原子速度 不需要校正,而系统动能在3/2NTkB上下10%浮动,此时 系统已经达到了热平衡。
在到达热平衡之前的轨迹信息和速度不需 要保存,因为那时的物理意义不严格,真 正对我们有意义的是达到平衡后的系统信 息。
• 通常的分子动力学模拟需要累积数百万步 以供分析,因而会产生极大的存储。
• 以1000个原子为例,储存每步的位置和速度约要10kB空间,则10000步需要100MB。
• 因为分子动力学计算的步长很短,每一步 移动的距离也很小,通常每隔10~20步存储 一次来节省硬盘空间。
NVE系综的 一般性分子 动力计算:
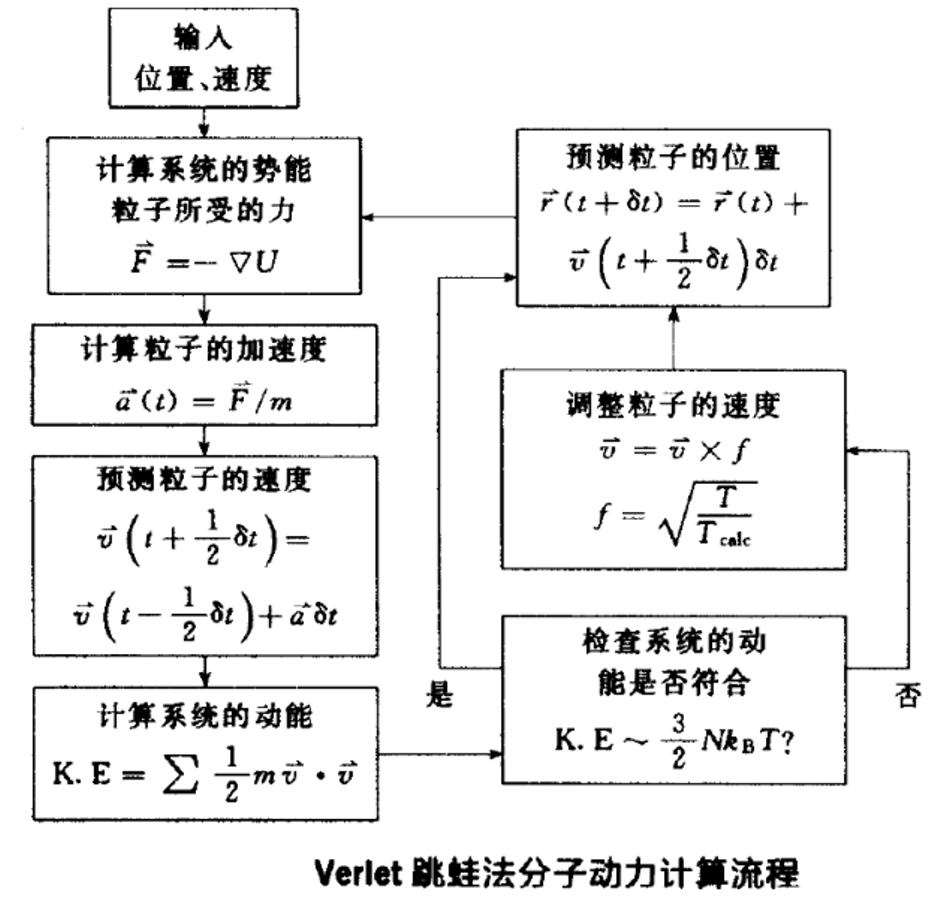
分子动力计算的初始设定:
执行分子动力学计算时,必须选择恰当的 初始条件,如恰当的初始温度、初始速度 、初始位置和积分步长等,如果初始条件 选择不当则会浪费很长的时间才能达到热 平衡,或者说根本达不到平衡。
初始结构:
初始结构的选取以越接近模拟体系的理想 结构越好。如要模拟体心立方的固体系统 时选取了面心立方作为初始结构,便是不 明智之举。
通常在模拟前作一些实验,降低系统的局 部高能区,使其接近于能量最低点的理想 结构,避免计算中由于偏离理想结构太多 而导致失败。
对于均匀相的液体,通常选用它的固相晶体结构作为初始模型,如果有XRD实验得 到的结构数据,则可以直接采用;如果没 有,通常采用均匀分布的面心立方结构排 列这些分子或原子。
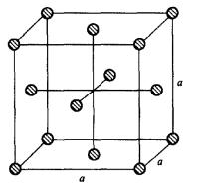
对于分子系统,通常将分子的质心放在面心立 方的格点上,分子的取向视系统而定。对于小分子可以随意指定其方向,如水和二氧 化碳分子等;
对于大分子则考虑取向时以不发生重叠为宜。
对于复杂液体,通常参考XRD或NMR实验 得到的结构位置,或者采用分子力学—— 能量最小化的结果作为起点。
初始速度
通常系统初始速度的产生,是先在-1~+1之间选取 一个呈高斯分布的随机数,再将其乘以系统所有 粒子的平均速度:
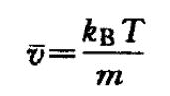
即可以得到符合麦克斯韦-玻尔兹曼分布的粒子速 度:
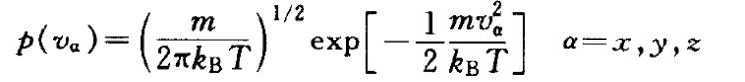
通常在实验前还要检查各方向动量是否为0,避免 计算过程中系统移动而导致能量不稳定。

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









