






| 具体内容 | |
| 模型 | scGPT,2024.2 |
| 数据类型 | 单细胞RNA测序数据 |
| 数据量 | 超过1000万个单细胞 |
| 算法 | GPT based,Flash-Attention,动态掩码 |
| 输入 | 细胞表达能力嵌入(分箱)+ 基因嵌入 + 条件标记;[cls] + 序列 + [pad] |
| 输出 | 预测基因表达值,学习细胞嵌入CLS |
| 分词方式 | 无传统分词 |
| 评估方式 | Cross-entropy(原始表达值,预测的掩码表达值) |
| 下游任务 | 细胞类型注释、扰动预测、多组学整合 |
| 创新方式 | 首次将生成式Transformer应用于单细胞领域,支持 “基因提示” 和 “细胞提示” 两种生成模式 |
尽管标记和提示的使用相似,但由于数据的非顺序性质,建模遗传读取本质上与自然语言不同。与句子中的单词不同,细胞内的基因顺序是可以互换的,并且没有等效的 “next gene” 概念可以预测。这使得将 GPT 模型的因果掩蔽公式直接应用于单细胞域变得具有挑战性。为了应对这一挑战,我们为 scGPT 开发了一种专门的注意力掩蔽机制,该机制根据注意力分数定义预测顺序。
!scGPT 的注意力掩码以统一的方式支持基因提示和细胞提示生成。二进制注意力掩码应用于 transformer 块中的自注意力图。对于 M 个词元的输入 hl(i)∈RM×D(参见在线方法 4.2.1),transformer 块将生成 M 查询和关键向量来计算注意力图,A∈RM×M。注意掩码的大小相同 M×M 。我们在补充图 S1A 中可视化了注意力掩码,其中查询按行组织,键按列组织。与掩码的每一列关联的令牌标识在图的底部注释,namely<cls> ,已知基因,以及
未知基因。输入嵌入 hl(i) 中的每个标记都可以是以下三组之一:(1)
保留的 <CLS> 用于细胞嵌入的标记(在在线方法 4.2.2 中引入),(2) 具有标记嵌入和表达值嵌入的已知基因,以及 (3) 要预测其表达值的未知基因。scGPT 的注意力掩盖的经验法则是只允许在 “已知基因” 的嵌入和查询基因本身之间进行注意力计算。在每次世代迭代中,scGPT 都会预测一组新基因的基因表达值,这些基因反过来又成为下一次迭代中的 “已知基因”,用于注意力计算。这种方法通过在非序列单单元数据中进行顺序预测,反映了传统 transformer 解码器中带有下一个标记预测的随意掩码设计。
如补充图 S1A 所示,在训练过程中,我们随机选择基因的比例为未知,因此在输入中省略了它们的表达值。对这些未知基因位置的查询只允许在对已知基因和查询基因本身进行注意力计算时进行。在每次迭代中,scGPT 从未知集中选择具有最高预测置信度的前 1/K 基因作为已知基因包含在下一次迭代 i+1 中。直观地说,该工作流程以自回归方式简化了大组基因表达的生成,其中首先生成具有最高预测置信度的基因表达,并用于帮助后续轮次生成。基因提示生成以迭代方式类似地工作。区别在于,它从一组具有观察到的表达值的已知基因开始,而不是细胞包埋。
scGPT 注意力掩蔽统一了已知基因的编码过程和未知基因的生成。它也是最早对非序列数据进行自回归生成的 transformer 方案之一。
Cls代表整个细胞的embedding
输入与数据格式
3. 输出
4. 数据量
5. 下游任务
6. 损失函数
7. 具体算法
8. 创新方式
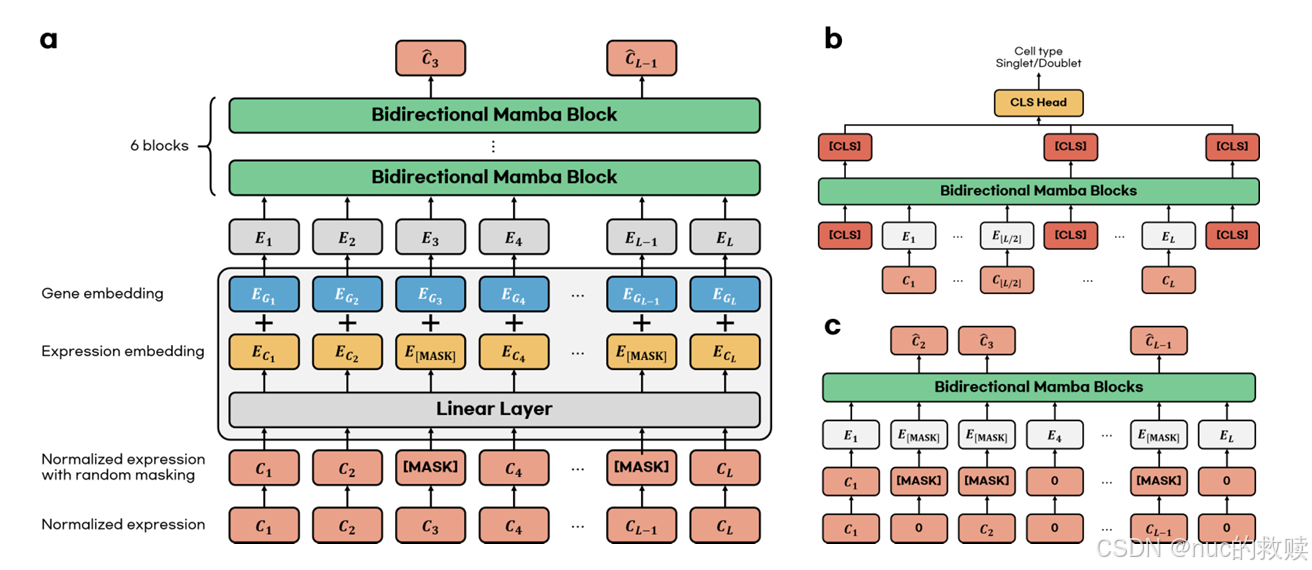
| 具体内容 | |
| 模型 | scMamba,2025.2 |
| 数据类型 | 单核RNA测序的原始基因表达矩阵,包含所有基因的数值 |
| 数据量 | 160万个细胞 |
| 算法 | 基于Mamba的改进模型,核心为Bidirectional SSM |
| 输入 | 细胞表达连续值嵌入 -mask 15% + 基因嵌入(gene2vec、保留所有基因) |
| 输出 | 预训练输出掩码预测值,微调输出根据下游任务不同而变化 |
| 分词方式 | 无传统分词 |
| 评估方式 | MSE(预测被掩码的连续数值) |
| 下游任务 | 细胞类型注释、双联体检测、插补预测、差异表达基因识别 |
| 创新方式 | Mamba模型首次应用于snRNA-seq分析,不降维,不高变基因选择 |
模型分析:scMamba
1. 发布时间
2. 输入与输出
3. 输入数据格式
4. 数据量
5. 分词方式
6. 下游任务
7. 预训练损失函数
8. 具体算法
9. 创新方式
| 具体内容 | |
| 模型 | scFoundation,2024.5 |
| 数据类型 | 单细胞RNA测序数据 |
| 数据量 | 超过5000万单细胞转录组数据 |
| 算法 | Transformer-based,12个encoder 12头,6个decoder 8头;不对称编码器-解码器 |
| 输入 | 细胞的基因表达向量通过嵌入模块将连续表达值映射为高维向量,未离散化--添加 T 和 S 值(RDA)-- 下采样 -- mask 30% 后输入 |
| 输出 | 细胞嵌入、基因上下文嵌入 |
| 分词方式 | 无传统分词 |
| 评估方式 | 回归损失(MAE/MSE) |
| 下游任务 | 基因表达增强、组织药物响应预测、单细胞药物响应分类、单细胞扰动预测等 |
| 创新方式 | 通过预训练模型不微调实现了多种下游任务表现的提升,消除测序深度带来的 Embedding 差异 |
Read-Depth-Aware 预训练任务
一个基因表达量越低,就越有可能因为测序深度的不足,而成为 dropout (0) 值的情况。在训练过程中,模型逐渐学会了通过其他基因的表达情况,去建模需要预测的基因真实的表达值,从而优雅地处理了 Dropout 问题,而且从计算层面提升了测序深度。
T 指的是下采样之前总的 counts 数,但从 get_embedding.py 来看,还经过了 log10 转换。
S 指的是下采用之后总的 counts 数,同样经过了 log10 转换。
这两个位置的信息告诉了 Encoder 需要消除测序深度带来的 Embedding 差异,将输入基因和细胞的 Embedding 表示映射到 Target 测序深度上去,能够:
获得没有测序深度差异的 Cell Embedding
Encoder 的 Gene Embedding 参与到 Decoder 还原基因表达量的过程中,同样实现了不受测序深度影响的目的
研究评估了 scFoundation 应用于多种下游任务的表现。值得一提的是,多数任务直接使用了未经微调的预训练 scFoundation 模型,大大降低了应用基座模型的门槛和成本:
a. 获得增强测序深度的基因表达 (即常见的 Imputation 任务)。scFoundation 超过了多个领域内流行的 Imputation 方法,且这些方法需要在特定数据上的额外训练,而 scFoundation 只是预训练模型
b. 癌症药物反应预测。将 DeepCDR 模型的 MLP 部分输出替换成了 scFoundation Encoder 得到的 Cell Embedding,新的 DeepCDR 在训练后取得了比原有模型更好的预测表现
c. 单细胞水平的药物反应分类预测。scFoundation 预训练模型 Eecoder 得到的 Cell Embedding 作为 SCAD 模型的输入进行训练,取得了比原有 SCAD 模型更好的表现
d.Perturb-Seq 细胞干扰试验结果预测。在 scFoundation 预训练模型 Decoder 输出的 Gene Embedding 的基础上,研究者建立了细胞特异的基因共表达 Graph,并利用此 Graph 替代了 GEAR 模型原有的 Graph,在干扰预测任务上取得了比原有 GEAR 模型更好的表现。
e. 其他下游任务:细胞类型注释、基因 Module 和基因调控网络的推理
| 具体内容 | |
| 模型 | SpliceBERT,2023.5 |
| 数据类型 | pre-mRNA序列 |
| 数据量 | 201万条序列,650亿核苷酸 |
| 算法 | BERT-based,6层,512维,16头;One-hot位置嵌入 |
| 输入 | 随机掩盖15%的核苷酸 |
| 输出 | 预测被掩盖的核苷酸 |
| 分词方式 | 单核苷酸分词,添加[CLS]、[SEP]、[MASK] |
| 评估方式 | Cross-entropy(原始值,预测的掩码值) |
| 下游任务 | 跨物种剪接位点预测,人类分支点预测,剪接变异影响预测 |
| 创新方式 | 首次在72种脊椎动物pre-mRNA序列上预训练,捕捉进化信息 |
不均一核RNA为存在于真核生物细胞核中的不稳定、大小不均的一组高分子RNA(分子量约为105~2×107,沉降系数约为30—100S)之总称。占细胞全部RNA之百分之几,在核内主要存在于核仁的外侧。
模型分析:SpliceBERT
1. 发布时间
2. 输入
3. 输出
4. 输入数据格式
5. 数据量
6. 分词方式
7. 下游任务
8. 预训练损失函数
9. 具体算法
10. 创新方式
| 具体内容 | |
| 模型 | RhoFold+,2024.12;预训练模型的使用 |
| 数据类型 | RNA序列 |
| 数据量 | 2370万RNA序列 |
| 算法 | RNA-FM生成序列嵌入 + MSA -- Rhoformer、几何感知注意力、IPA模块 |
| 输入 | RNA序列、多序列比对文件 |
| 输出 | 3D结构PDB、二级结构dotbracket |
| 分词方式 | 单核苷酸分词 |
| 评估方式 | 坐标均方误差MSE、几何约束损失、不变点注意力IPA损失、交叉熵损失 |
| 下游任务 | 单链RNA 3D结构预测、二级结构预测、螺旋间角度预测、结构质量评估 |
| 创新方式 | 首次将预训练语言模型用于RNA结构预测 |
RhoFold+模型分析
输入
输出
数据格式
下游任务
损失函数
具体算法
创新方式
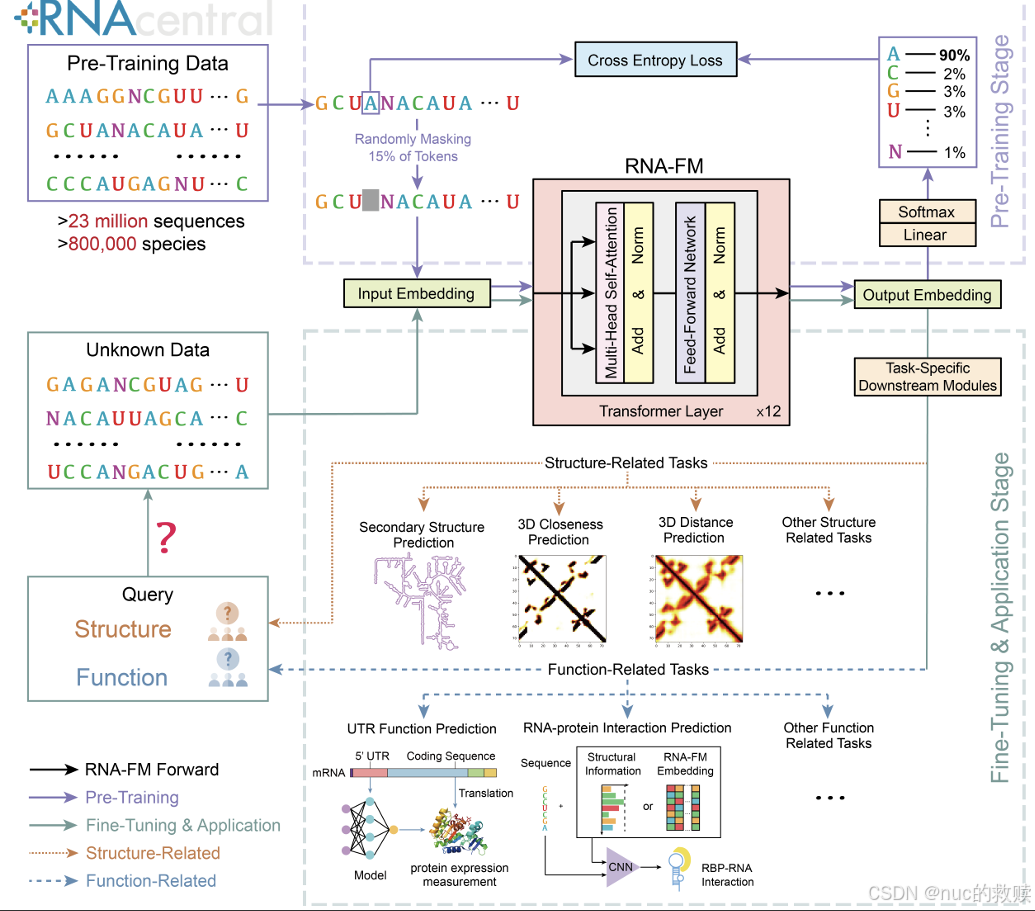
|
具体内容
| |
|
模型
|
RNA-FM,2022.4
|
|
数据类型
|
非编码 RNA
|
|
数据量
|
2370 万个非冗余 RNA 序列,大规模跨物种序列
|
|
算法
|
BERT-based;12层encode,隐藏层维度640,每层包含20头;
|
|
输入
|
MLM,
多级掩码输入序列中15%-20%的token,[CLS] + 序列 + [SEP]
|
|
输出
|
640维的CLS全局序列表示和token的embedding
|
|
分词方式
|
支持
单核苷酸分词、k-mer分词、密码子分词
|
|
评估方式
|
多级
MLM
损失,单碱基、子序列级、motif级掩码的加权交叉熵
|
|
下游任务
|
RNA二级/三级结构预测、RNA功能预测、RNA分类、mRNA相关任务、SARS-CoV-2 基因组结构和进化预测、蛋白质-RNA 结合
|
|
创新方式
|
首个通用目的RNA语言模型
|
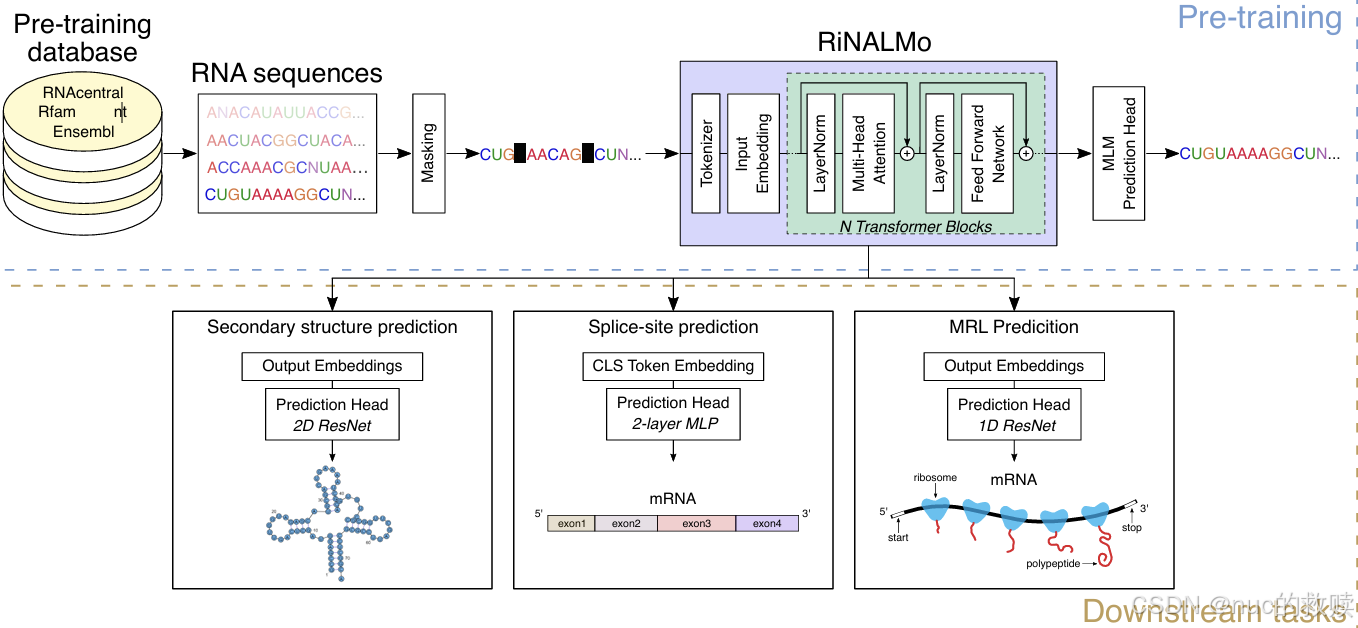
|
具体内容
| |
|
模型
|
RiNALMo
,
2024.3
|
|
数据类型
|
非编码
RNA
|
|
数据量
|
3600
万个非冗余
RNA
序列
|
|
算法
|
BERT-based
;
33
层
encode
,每层包含
20
头;
RoPE
位置编码;
SwiGLU
激活
|
|
输入
|
MLM
,随机掩码输入序列中
15%-20%
的
token
,
[CLS] +
序列
+ [SEP]
|
|
输出
|
1280
维的
CLS
全局序列表示和
t
oken
的
embedding
|
|
分词方式
|
支持
单核苷酸分词
、
k-
mer
分词、密码子分词
|
|
评估方式
|
Cross-entropy(
原始序列,预测的掩码序列
)
|
|
下游任务
|
RNA
序列特征提取、序列和核苷酸级别的分类回归任务,以及
RNA
接触预测
|
|
创新方式
|
对特殊核苷酸采用独立
token
编码
引入
FlashAttention-2
算法降低显存消耗,支持超长序列处理
|
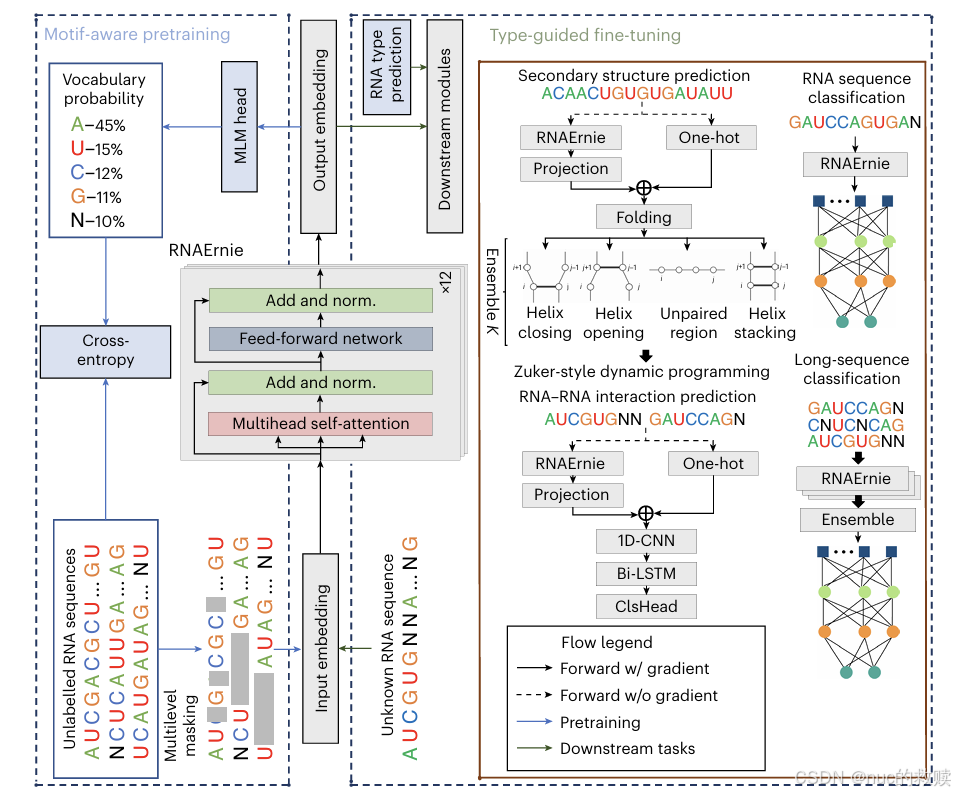
|
具体内容
| |
|
模型
|
RNAErnie
,
2024.5
|
|
数据类型
|
非编码
RNA
|
|
数据量
|
2300
万个非冗余
RNA
序列
|
|
算法
|
BERT-based
;
12
层
encode
,隐藏层维度
768
,
12
头;
RoPE
位置编码
|
|
输入
|
MLM
,
多级掩码
输入序列中
15%-20%
的
token
,
[CLS] +
序列
+
[
IND]
|
|
输出
|
768
维的
CLS
全局序列表示和
t
oken
的
embedding
|
|
分词方式
|
支持
单核苷酸分词
、
k-
mer
分词、密码子分词
|
|
评估方式
|
多级
MLM
损失
,单碱基、子序列级、
motif
级掩码的加权交叉熵
|
|
下游任务
|
RNA
序列分类准确率
98.7%
、
RNA-RNA
相互作用和
RNA
二级结构预测
F1=0.82
|
|
创新方式
|
预训练阶段采用:碱基级掩码、子序列级掩码和
motif
随机掩码
针对下游任务的类型引导微调:在序列末尾
附加
RNA
类型标记
|
|
具体内容
| |
|
模型
|
UNI-RNA,2023.07
|
|
数据类型
|
RNA序列
|
|
数据量
|
约10亿条RNA序列,MMseqs2聚类
|
|
算法
|
BERT-based;24层encode,隐藏层维度1280,12头;RoPE位置编码
|
|
输入
|
MLM,
多级掩码输入序列中15%-20%的token,[CLS] + 序列 + [SEP]
|
|
输出
|
768维的CLS全局序列表示和token的embedding
|
|
分词方式
|
单核苷酸分词
|
|
评估方式
|
Cross-entropy(原始序列,预测的掩码序列)
|
|
下游任务
|
二级结构预测、三级接触图预测、5‘UTR的ribosome load预测、跨物种剪接位点预测、ncRNA家族分类、RNA修饰位点预测
|
|
创新方式
|
支持长序列4096长度,结合FlashAttention、RoPE、fused layernorm
|

FlashAttention的核心原理是通过将输入分块并在每个块上执行注意力操作,从而减少对高带宽内存(HBM)的读写操作
Rope:旋转位置编码,目前是大模型相对位置编码中应用最广的方式之一。
1. 输入
2. 输出
3. 数据格式
4. 分词方式
5. 下游任务
6. 损失函数
7. 具体算法
8. 创新方式
9. 潜在挑战
|
具体内容
| |
|
模型
|
GenerRNA
|
|
数据类型
|
RNA
核苷酸序列
|
|
数据量
|
1609
万条去重
RNA
序列
|
|
算法
|
GPT-2 based
;
24
层,
1280
维
|
|
输入
|
经过
BPE
分词后的
token
序列
|
|
输出
|
自回归生成的
RNA token
序列,解码后为核苷酸序列
|
|
分词方式
|
Byte-Pair Encoding
,词汇表大小是
1024
|
|
评估方式
|
自回归预测每个
token
的条件概率的负对数似然之和
|
|
下游任务
|
生成具有稳定二级结构的新
RNA
、生成特定蛋白的高亲和力
RNA
。
|
|
创新方式
|
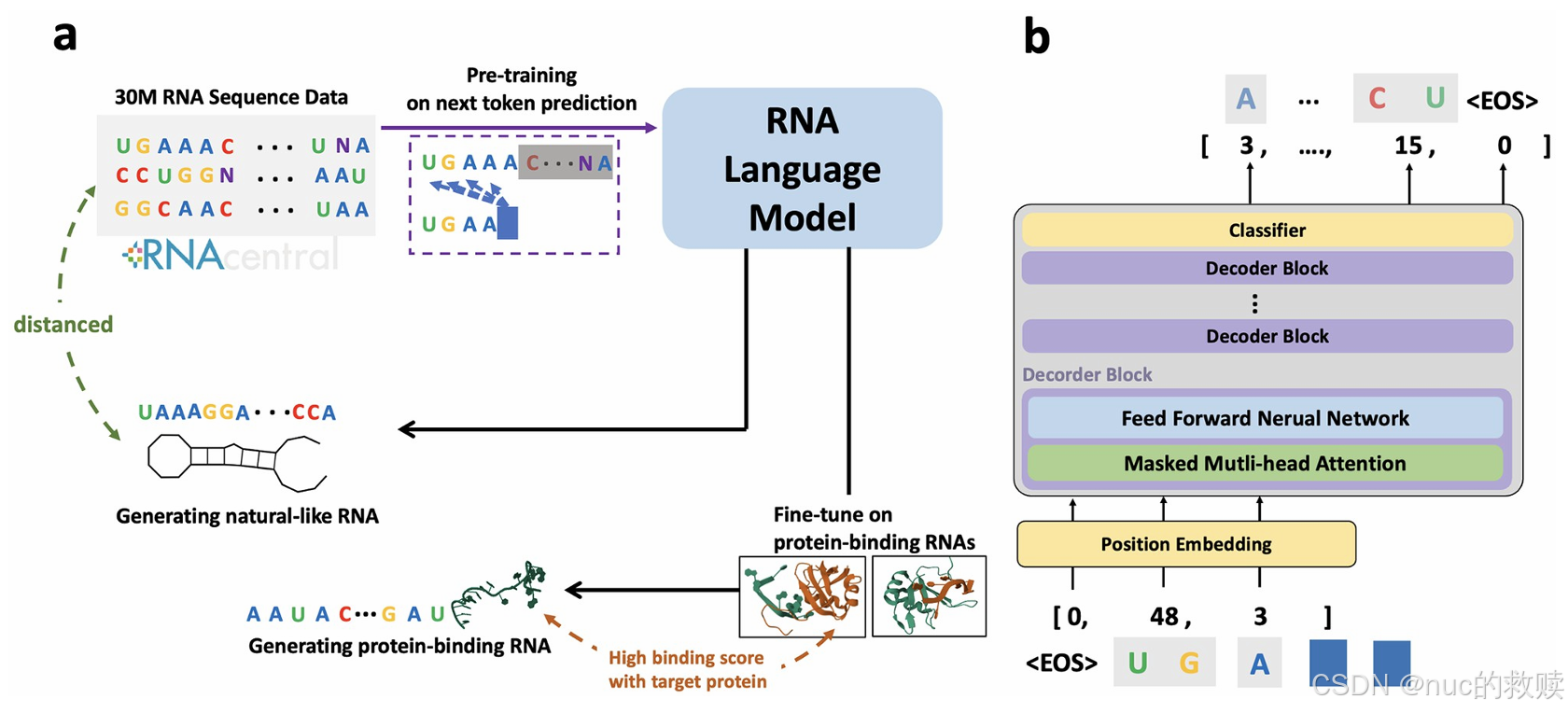
分词方式明确使用了BPE,可能合并常见的核苷酸组合成单个token,词汇表大小是1024。BPE在训练分词器时处理了100万条RNA序列,尝试了不同词汇量,最终选择1024,因为平衡了信息压缩和模型处理能力。BPE的优势在于能处理可变长度的子词,相比k-mer或单核苷酸分词,能更有效地压缩信息,处理更长序列。
使用Adam优化器,学习率预热到1e-3后衰减到1e-4。生成时采用不同的采样策略,如贪心搜索、束搜索和随机采样(top-k),其中随机采样在生成自然分布RNA时效果最好。
Eos代表每个序列的起始和终点。
预训练使用的是负对数似然(NLL),公式是NLL_seq = -Σ log pθ(xi | x<i),即自回归预测每个token的条件概率的负对数似然之和。
下游任务评估方法创新:结合MFE(最小自由能)稳定性分析、同源性搜索(nhmmer)和k-mer分布KL散度,全面验证生成质量。
稳定性验证:生成的RNA MFE接近天然序列,显著优于随机/打乱序列。
1. 输入与输出
2. 数据格式
3. 分词方式
4. 下游任务
5. 损失函数
NLL seq =− ∑ i =1 L log pθ ( xi ∣ x < i )
6. 具体算法
7. 创新点
关键细节补充
|
具体内容
| |
|
模型
|
scBERT
,
2021.12
|
|
数据类型
|
单细胞
RNA
测序数据,基因表达谱被转换成
基因嵌入和表达嵌入
|
|
数据量
|
200
万单细胞
RNA
测序
数据
|
|
算法
|
BERT-based
(
Performer
);
6
层
encode
,
10
头;无位置编码
|
|
输入
|
细胞表达能力分箱的嵌入
-mask 15% +
基因嵌入(
gene2vec
、
保留所有基因
)
|
|
输出
|
细胞类型的概率分布
|
|
分词方式
|
连续表达值被分箱(
binning
)为离散的区间,映射为
200
维的“表达嵌入”
|
|
评估方式
|
Cross-entropy(
原始序列,预测的掩码序列
)
|
|
下游任务
|
细胞类型注释、基因重要性分析、细胞表示学习
|
|
创新方式
|
首次
BERT
架构迁移至
scRNA
-seq
分析、采用
Performer
|
|
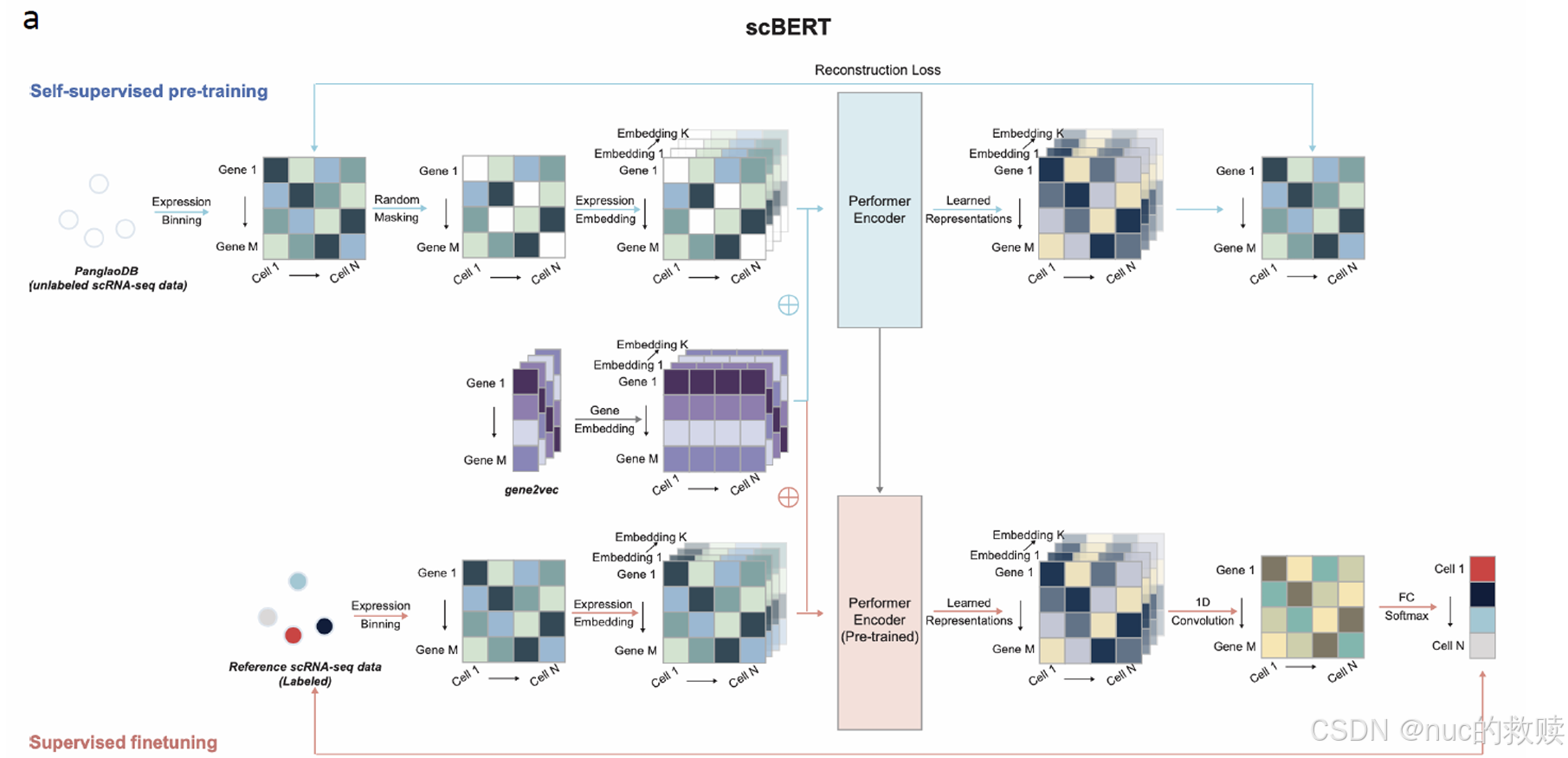
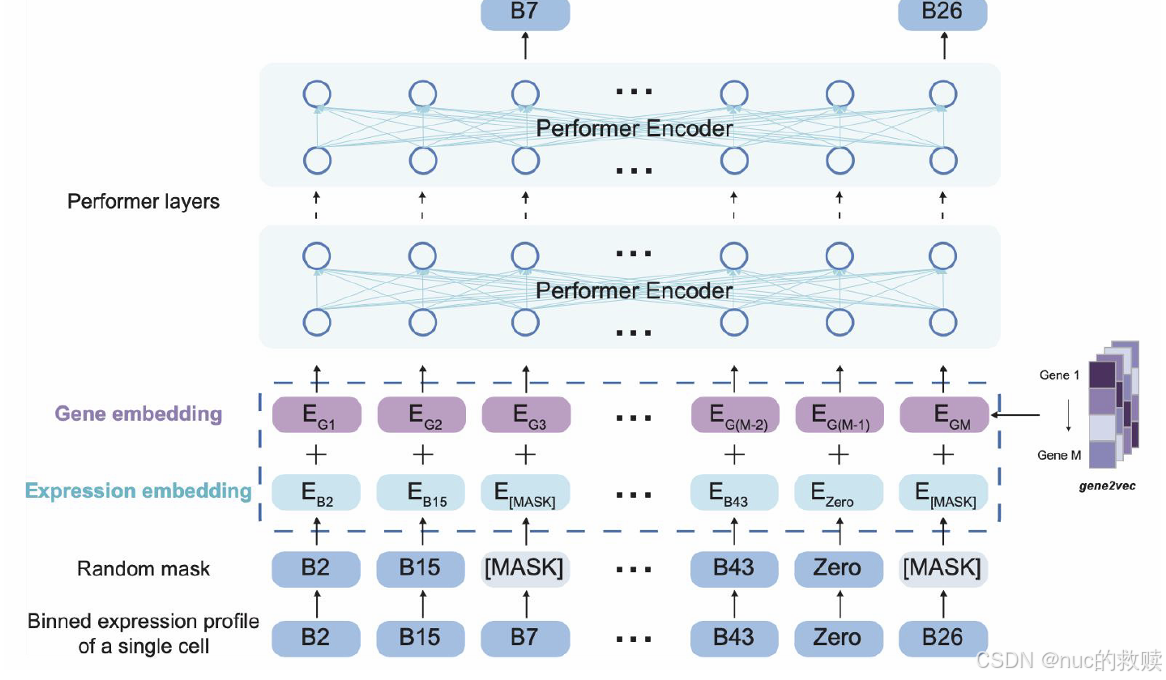
具体内容
| |
|
模型
|
scBERT,2021.12
|
|
数据类型
|
单细胞RNA测序数据,基因表达谱被转换成
基因嵌入和表达嵌入
|
|
数据量
|
112万单细胞RNA测序数据
|
|
算法
|
BERT-based(
Performer);6层encode,10头;无位置编码
|
|
输入
|
细胞表达能力嵌入(分箱) -mask 15% + 基因嵌入(gene2vec、
保留所有基因)
|
|
输出
|
细胞类型的概率分布
|
|
分词方式
|
单基因分词
|
|
评估方式
|
Cross-entropy(原始表达值,预测的掩码表达值)
|
|
下游任务
|
细胞类型注释、基因重要性分析、细胞表示学习
|
|
创新方式
|
首次BERT架构迁移至scRNA-seq分析、采用Performer
|
|
具体内容
| |
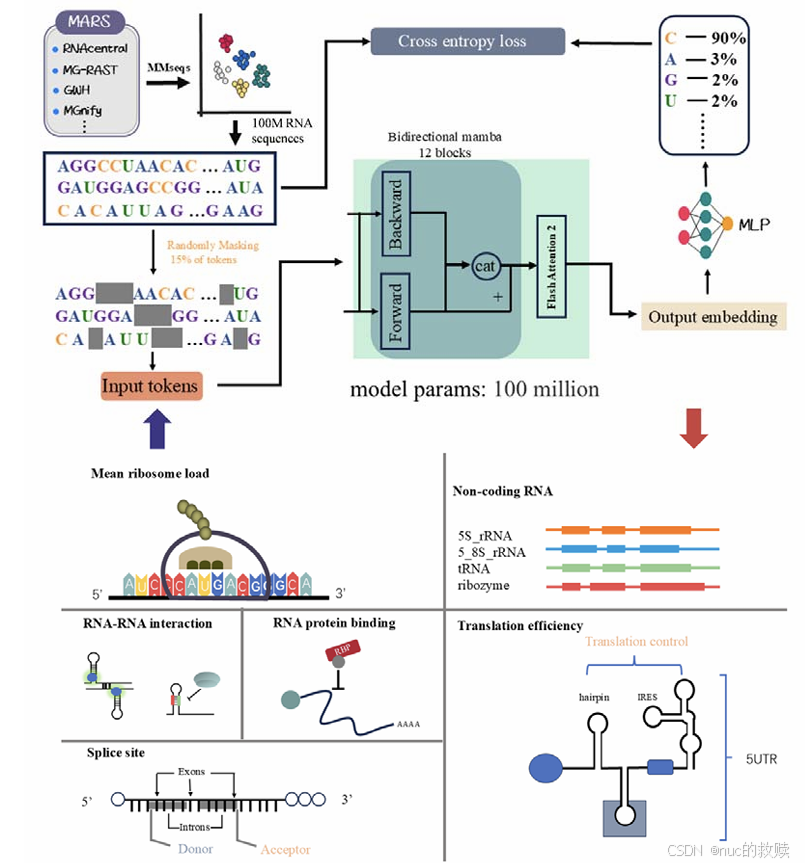
|
模型
|
DGRNA,2024.12
|
|
数据类型
|
RNA序列
|
|
数据量
|
约1亿条RNA序列,Mmseqs去冗余
|
|
算法
|
基于双向Mamba2模块(12层)+ Flash Attention-2层。
|
|
输入
|
MLM,随机掩码输入序列中15%的token,[CLS] + 序列 + [EOS]
|
|
输出
|
每个位置输出768维嵌入向量,或下游任务相关的结果
|
|
分词方式
|
单核苷酸分词
|
|
评估方式
|
Cross-entropy(原始表达值,预测的掩码表达值)
|
|
下游任务
|
非编码RNA分类、RNA-RNA/RNA-蛋白相互作用预测、剪接位点预测、翻译效率预测、RNA蛋白结合位点识别
|
|
创新方式
|
Mamba2架构+双向SSM+Flash Attention,6类下游任务中达到SOTA
|
|
具体内容
| |
|
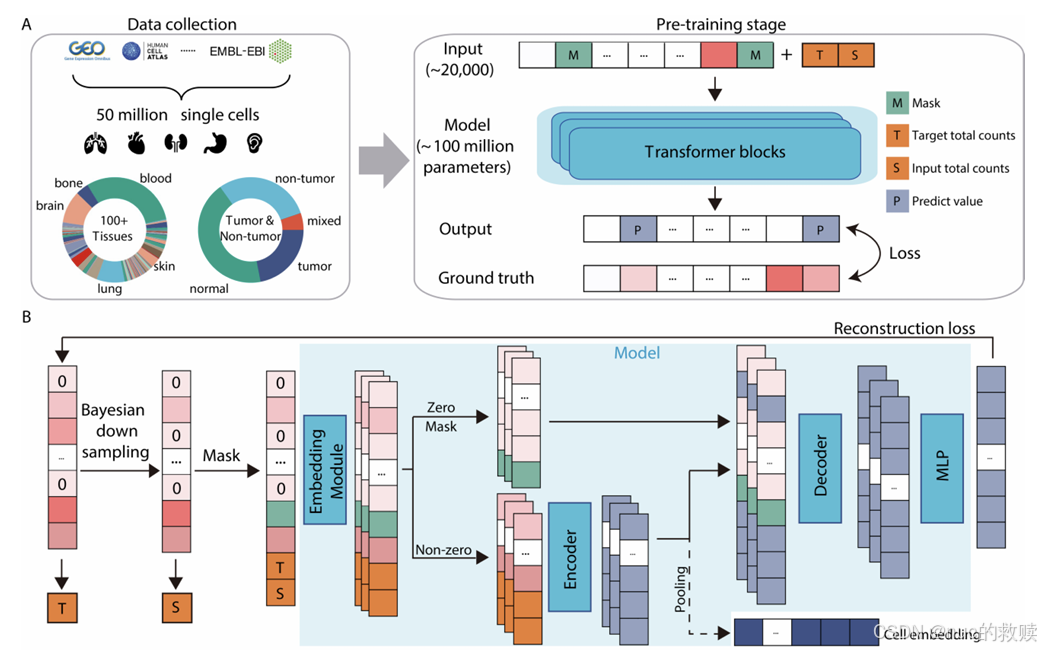
模型
|
scGPT,2024.2
|
|
数据类型
|
单细胞RNA测序数据
|
|
数据量
|
超过1000万个单细胞
|
|
算法
|
GPT based,Flash-Attention
|
|
输入
|
细胞表达能力嵌入(分箱)+ 基因嵌入 + 条件标记;[cls] + 序列 + [pad]
|
|
输出
|
预测基因表达值,学习细胞嵌入CLS
|
|
分词方式
|
单基因分词
|
|
评估方式
|
Cross-entropy(原始表达值,预测的掩码表达值)
|
|
下游任务
|
细胞类型注释、
扰动预测、
多组学整合
|
|
创新方式
|
首次将生成式Transformer应用于单细胞领域,支持 “基因提示” 和 “细胞提示” 两种生成模式
|
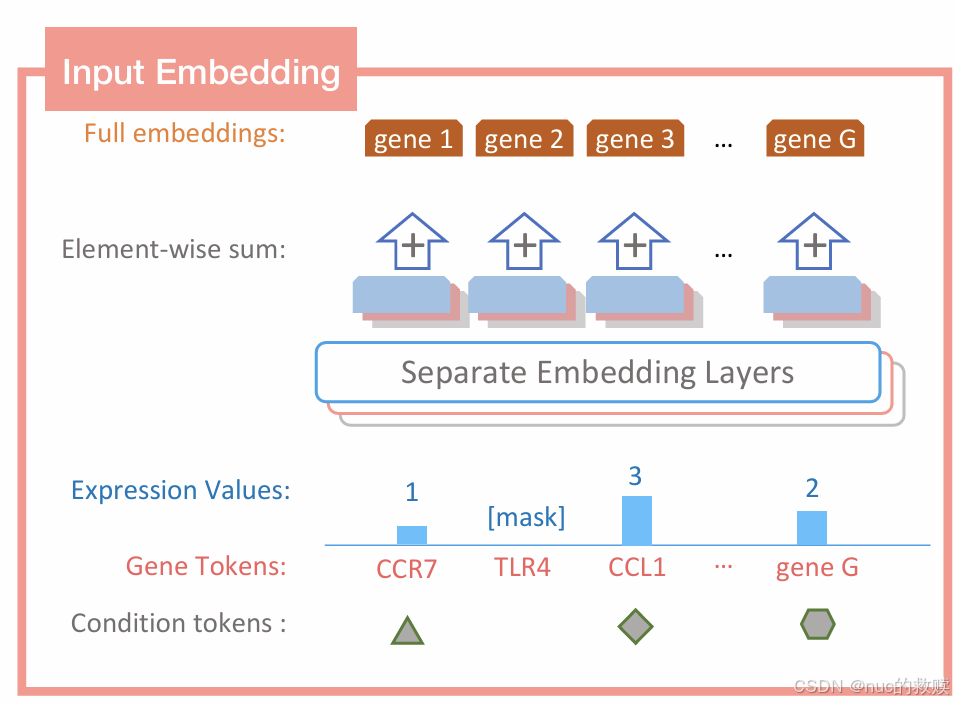 !!!!基因 表达 叠加
!!!!基因 表达 叠加
|
具体内容
| |
|
模型
|
MRM-BERT,2024.2
|
|
数据类型
|
长度为101的RNA序列片段
|
|
数据量
|
超30万
|
|
算法
|
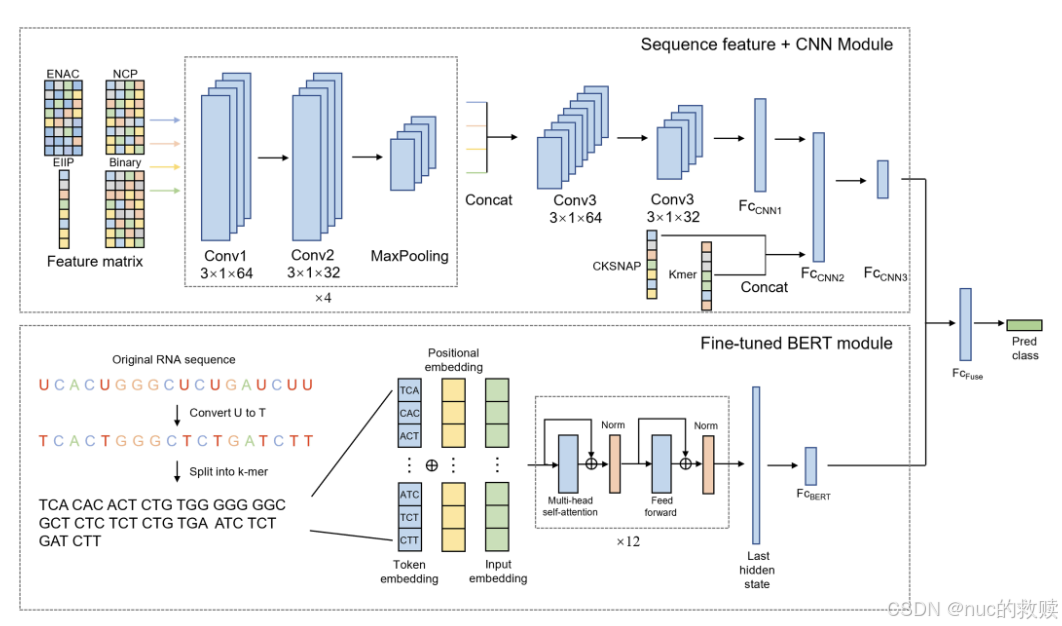
Fine-tuned DNABERT + CNN,二者输出拼接后经全连接层分类
|
|
输入
|
BERT
输入:3-mer分词后的索引序列
CNN
输入:多特征编码矩阵(通过其他现有method)
|
|
输出
|
中心位点是否为12种RNA修饰类型的概率
|
|
分词方式
|
3-mer滑动窗口分词
|
|
评估方式
|
交叉熵损失(分类任务)
|
|
下游任务
|
多任务RNA修饰位点预测(12种修饰类型的分类)
|
|
创新方式
|
首次结合预训练BERT表示与传统序列特征
|
|
具体内容
| |
|
模型
|
CodonBERT,2024.6
|
|
数据类型
|
mRNA的编码区序列(起始密码子—终止密码子)
|
|
数据量
|
超1000万条mRNA序列,覆盖哺乳动物、细菌、人类病毒、酵母等生物
|
|
算法
|
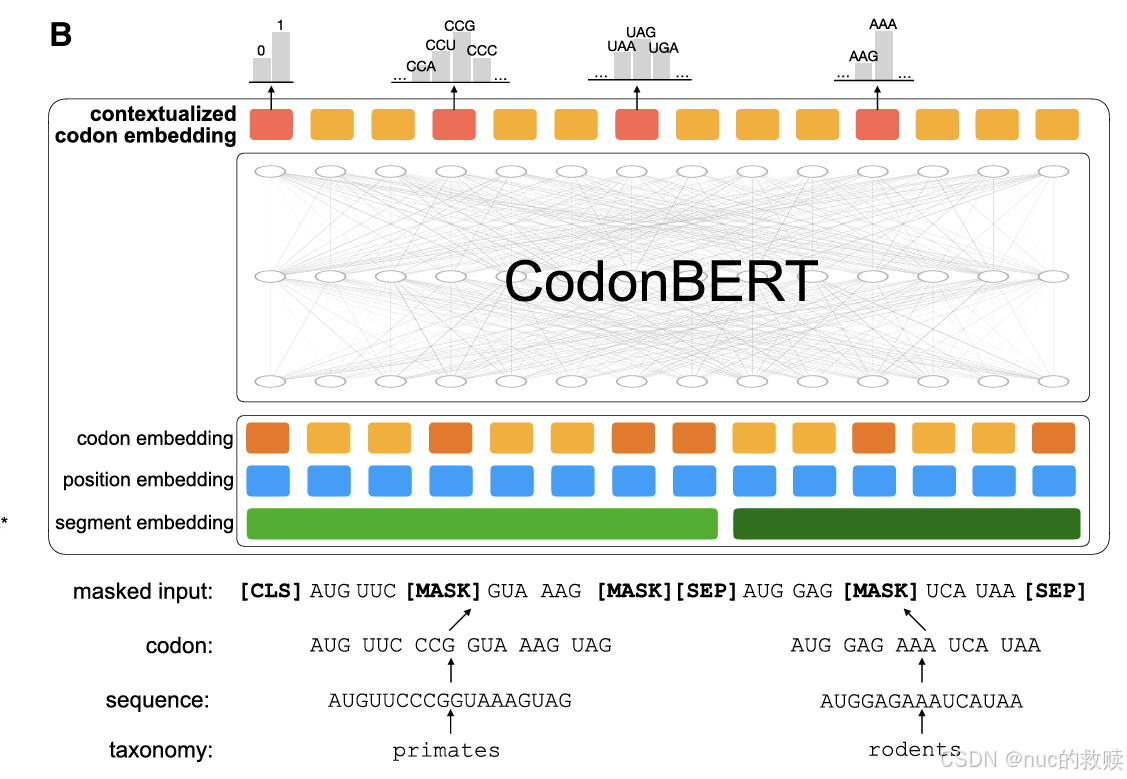
BERT-based,12层,12头,隐藏层维度768
|
|
输入
|
组合密码子嵌入、位置嵌入、序列嵌入,随机掩码15%的密码子;[CLS]、[SEP]、[UNK]、[PAD]、[MASK]特殊编码
|
|
输出
|
预测掩码位置的密码子(MLM)及序列对的分类群关系(STP)
|
|
分词方式
|
密码子分词
|
|
评估方式
|
MLM:交叉熵损失,预测被掩码的密码子
STP:交叉熵损失,判断两序列是否属于同一分类群
|
|
下游任务
|
蛋白质表达水平、mRNA降解速率、稳定性预测、蛋白质表达分类、疫苗抗原表达预测
|
|
创新方式
|
以密码子为基本单元,有两个预训练任务
|
|
具体内容
| |
|
模型
|
RNA-MSM,2024.4
|
|
数据类型
|
RNA多序列比对
|
|
数据量
|
约860万条序列,3932个Rfam家族
|
|
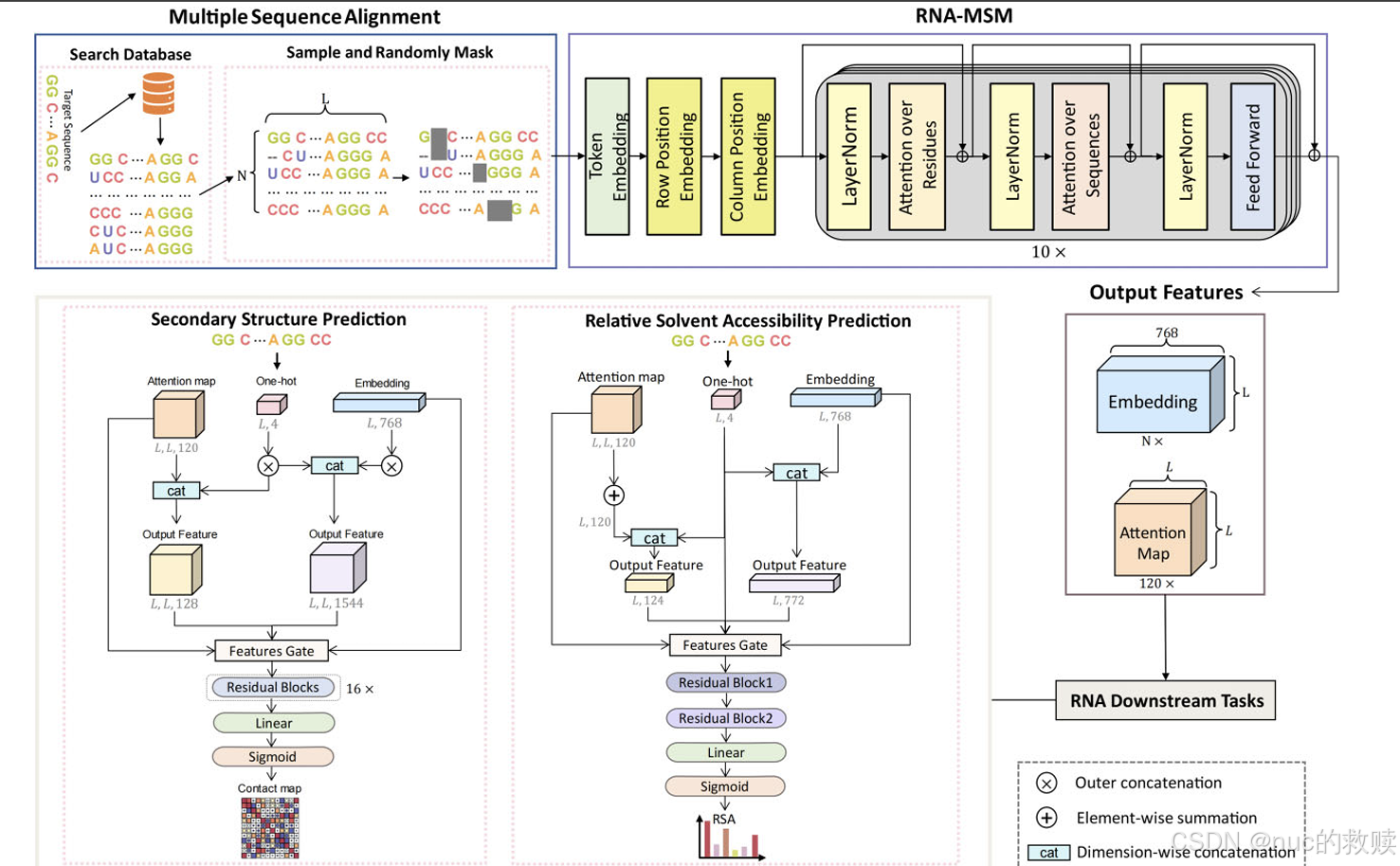
算法
|
改进自蛋白质MSA Transformer,10层,12头,768维
|
|
输入
|
MLM,MSA(N*L)随机mask 20% token。非标准核苷酸统一为X,gap保留为-
|
|
输出
|
2D
注意力图(L×L×120) :包含碱基配对概率信息
1D
嵌入表示(N×L×768):包含溶剂可及性等结构信息
|
|
分词方式
|
单核苷酸分词,词汇表包含6个字符
|
|
评估方式
|
Cross-entropy(原始表达值,预测的掩码表达值)
|
|
下游任务
|
RNA
二级结构预测,溶剂可及性预测
|
|
创新方式
|
直接利用多序列比对信息
|
|
具体内容
| |
|
模型
|
RNA-MSM,2024.4
|
|
数据类型
|
RNA多序列比对
|
|
数据量
|
约860万条序列,3932个Rfam家族
|
|
算法
|
改进自蛋白质MSA Transformer,10层,12头,768维
|
|
输入
|
MLM,MSA(N*L)随机mask 20% token。非标准核苷酸统一为X,gap保留为-
|
|
输出
|
2D注意力图(L×L×120) :包含碱基配对概率信息
1D嵌入表示(N×L×768):包含溶剂可及性等结构信息
|
|
分词方式
|
单核苷酸分词,词汇表包含6个字符
|
|
评估方式
|
Cross-entropy(原始表达值,预测的掩码表达值)
|
|
下游任务
|
RNA二级结构预测,溶剂可及性预测
|
|
创新方式
|
直接利用多序列比对信息
|
|
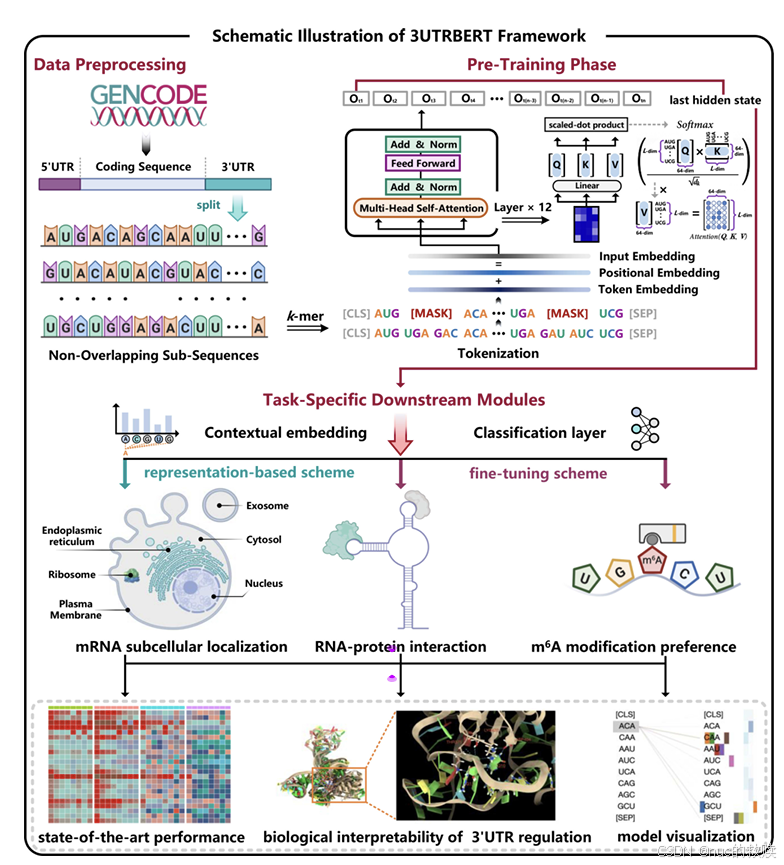
具体内容
| |
|
模型
|
3UTRBERT,2023.9
|
|
数据类型
|
mRNA的3'UTR序列片段.长度510核苷酸
|
|
数据量
|
20362条非冗余3'UTR序列,总计76435649核苷酸
|
|
算法
|
BERT-based,12层,12头,768维
|
|
输入
|
MLM,分词后mask 15%
|
|
输出
|
预测被遮盖k-mer的原始核苷酸组合
|
|
分词方式
|
k-mer分词,k=3
|
|
评估方式
|
Cross-entropy(原始值,预测的掩码值)
|
|
下游任务
|
RBP结合位点预测、m6A修饰位点预测、mRNA亚细胞定位
|
|
创新方式
|
首次应用
BERT
于
3'UTR
|
| 具体内容 | |
| 模型 | RhoFold+,2024.12;预训练模型的使用 |
| 数据类型 | RNA序列 |
| 数据量 | 2370万RNA序列 |
| 算法 | RNA-FM生成序列嵌入 + MSA -- Rhoformer、几何感知注意力、IPA模块 |
| 输入 | RNA序列、多序列比对文件 |
| 输出 | 3D结构PDB、二级结构dotbracket |
| 分词方式 | 单核苷酸分词 |
| 评估方式 | 坐标均方误差MSE、几何约束损失、不变点注意力IPA损失、交叉熵损失 |
| 下游任务 | 单链RNA 3D结构预测、二级结构预测、螺旋间角度预测、结构质量评估 |
| 创新方式 | 首次将预训练语言模型用于RNA结构预测 |
RhoFold+模型分析
输入
输出
数据格式
下游任务
损失函数
具体算法
创新方式

















 204
204

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









