






论文:Host genetic regulation of human gut microbial structural variation
杂志:Nature
年份:2024
研究动机:尽管宿主遗传对于肠道微生物多样性以及一些关键细菌丰度的影响已经被证明,然而宿主遗传对肠道菌群遗传多样性的影响仍然有待研究。
研究内容:对4个来自荷兰的队列的9,015名个体的人类遗传变异和肠道微生物结构变异之间的关联进行了荟萃分析。
研究结果:
1.在分泌以N-乙酰半乳糖胺(GalNAc)为终点的A型寡糖抗原的个体中(这一特征是由人类ABO和FUT2基因型共同决定),Faecalibacterium prausnitzii中含有GalNAc利用基因簇(GalNAc utilization gene cluster)的结构变异片段的存在率更高。这一发现在来自坦桑尼亚的队列中也得到了验证。
2.体外实验表明,GalNAc可以作为携带GalNAc代谢途径的F. prausnitzii菌株的唯一碳水化合物来源。
3.进一步的计算和体外研究表明,其他ABO相关物种也可以利用GalNAc,特别是Collinsella aerofaciens.
4.GalNAc利用基因还与宿主的心血管代谢健康相关,特别是在携带黏膜A抗原的个体中。
综上所述,本研究表明,人类基因组和细菌宏基因组的遗传关联可以为细菌-微生物组相互作用提供新的见解。
相关背景介绍
细菌基因组的变异可以导致细菌菌株在适应性、碳水化合物利用、代谢能力、致病性以及其他生物学特性方面存在差异。细菌结构变异(SV)是高度可变的基因组片段,长度可变,可以对微生物的功能产生显著的影响,增加细菌的可塑性并帮助其快速适应环境。SV在人类肠道微生物基因组毕竟常见,并且人类个体之间的微生物SV存在很大的个体差异。基于宏基因组测序鉴定出来的缺失SV(dSV:在宏基因组样本中可检测到或不存在的基因组区域)和可变SV(vSV:丰富度在样本中高度可变的基因组区域)揭示了肠道微生物SVs与人类健康的关联。
纵向分析表明,肠道微生物SVs显示出物种特异的时间稳定性,这表明了肠道细菌对个体特异肠道环境的潜在适应。然而,目前对于人类基因如何塑造个体肠道环境,并对肠道微生物的遗传景观施加选择性压力的了解还很有限。
因此,在本研究中,我们对人类基因型与肠道微生物中的微生物SV之间的遗传关联进行了大规模的荟萃分析,涉及了四个来自荷兰队列的9015个个体。然后,在坦桑尼亚队列中对找到的关联进行了验证。
研究结果
1.肠道微生物SV的GWAS分析工作流程
数据:9015个荷兰个体,宏基因组和宿主遗传数据均可获得。分别来自四个荷兰队列:荷兰微生物组项目(DMP: n = 7,372), Lifelines-DEEP (LLD, n = 981), 500功能基因组项目(500FG,n = 396)以及300-Obesity (300OB; n = 266);本研究还引入了来坦桑尼亚队列(300TZGG, n = 279)作为验证队列,以验证在具有不同遗传背景和生活方式的个体中是否也具有相似的结论。

图1a 肠道微生物结构变异的GWAS分析流程。
基于宏基因组测序生成肠道微生物SV谱,然后将检测到的dSV和vSV和人类基因组中超过600万个常见的SNP基因型进行关联分析。遗传关联结果以vSV abundance在不同基因型的箱线图、dSV的柱状图展示以及全基因组水平的曼哈顿图的形式展现。
微生物SV的检测:基于SGV-Finder工具(https://github.com/segalab/SGVFinder)。简而言之,该算法通过将宏基因组测序短读段映射到参考基因组上,并解决了模糊比对的问题,然后和对微生物基因组进行分割成箱bin(每1kb的片段)。然后跨样本比较这些箱的宏基因组覆盖率。SGV-Finder将25-75%样本中覆盖率接近0的bin识别为dSV,将覆盖率高度可变的bin识别为vSV。这里SV的鉴定仅适用于具有足够宏基因组测序覆盖度的肠道微生物物种。
SGV-Finder主要包括两个关键步骤: 1. 基于迭代覆盖的读段分配算法(iterative-coverage-based read assignment algorithm)解决多重比对的模糊读段问题。即将模糊比对的宏基因组测序读段进行迭代重新分配给最有可能的参考基因组。2. 将每个参考基因组进行分割片段成为基因组箱(genomic bins),并跨样本检测这些基因组箱的覆盖度。对于dSV的确认:将覆盖度在25%-75%样本中都接近0的片段视为dSV。若存在共现率高度相关的dSV则被合并到更大的SV区域中。对于vSV的确定:使用Z-score方法标准化每个样本内物种基因组的覆盖度,然后对所有样本的每个基因组箱继续评估。将那些基于β′分布高度可变的视为vSV。进一步地将高度相关的vSV合并到更大的SV区域中。
这里SV检测中所使用的参考基因组是SGV-Finder所提供的参考基因组数据库,该数据库是基于proGenomes数据库(http://progenomes1.embl.de/)生成的。
SV检测结果:在108个肠道微生物物种中检测到了14,196个SV,包括10265个dSV和3931个vSV,平均每个物种具有3-379个SV。SV数量最多的物种是Dorea formicigenerans、Dorea Iongicatena和Blautia wexlerae. 对于不同物种来说,具有足够覆盖度检测SV的样本数量从11到7716不等。这些物种的平均丰富度占据总微生物组组成的80%。
为了确保与宿主遗传关联分析的统计效力,本研究选择了在至少10%样本中检测到的vSV和缺失率在5%-95%之间的dSV。同时,只保留了至少在两个队列中存在的SV。因此,总共纳入进去了共3552个SVs, 包括来自49个细菌物种的1666 dSVs和1886 vSVs(图1b, c)。对于遗传度和关联分析,对于vSV的覆盖度值采用基于秩的逆正态变换(inverse normal rank transformation)进行归一化处理。
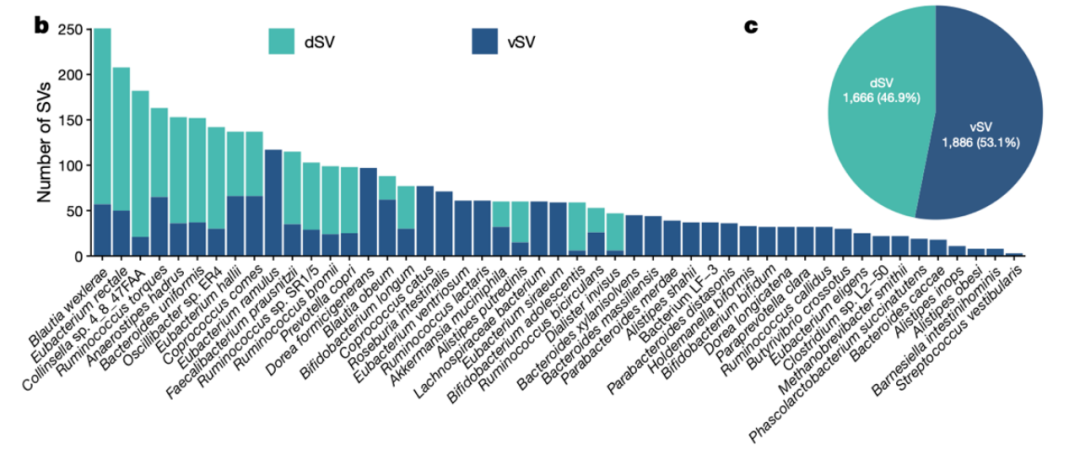
图1b-c 纳入GWAS分析的49个细菌物种上检测到的SV的数量统计
2.肠道微生物SV的遗传力(Heritability)
为评估肠道微生物SV在多大程度上会受到宿主遗传的影响,这里首先计算了DMP队列的1092个一级或二级亲缘关系对中出现的1339个SVs中遗传度(1339/3552 SVs)。
在基于物种丰度校正后,基于family水平的遗传度(h2)估计结果显示一个可遗传的dSV展现出显著的遗传度(FDR < 0.05): 该dSV是位于F. prausnitzii的一个2kb的dSV (577 – 579 bp), 其h2值为0.38。此外,还有26个dSVs和51个vSVs展示出名义上显著的遗传度(未经过多重校正,p < 0.05),其平均遗传度大小分别是0.28和0.41。
接下来则是进一步比较了不同物种其丰富度的遗传度以及SV的遗传度,观察到宿主遗传对于微生物的SV具有额外的效应(附图2)。(注意:本研究仍然缺乏足够的能力进行遗产度的计算和比较,对于物种丰富度和结构变异的精确遗传度的计算需要更大的样本量以及谨慎的实验设计。)
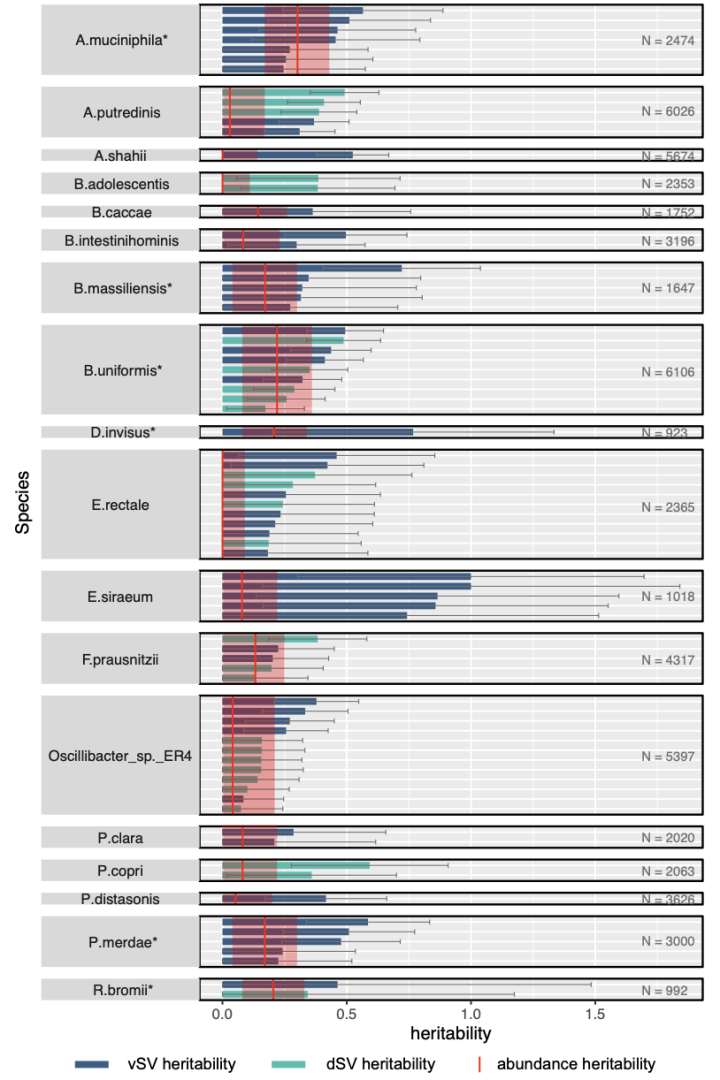
附图2 肠道微生物SV的遗传力以及相应的物种丰度的遗传力。
带*的物种表示具有显著丰富度遗传力的物种。只展示了p < 0.05遗传力显著的SV。
3.ABO基因座与F. prausnitzii的SVs
对每个队列的3552个SVs以及超过六百万人类SNPs进行关联分析,并进行荟萃分析。
结果显示,在经过Bonferroni校正的P < 0.05水平下显著的遗传关联几乎全是ABO位点与F. prausnitzii的SVs的关联,包括4个dSV和1个vSV(图2)。其中最强的关联是发现在rs635634与F. prausnitzii的一个2kb的dSV区域(577-579kb)之间(bmeta = 0.88, Pmeta = 1.21 x 10-45)。SNP rs635634是位于ABO基因中,该基因编码一个糖基转移酶,可以修饰细胞表面的寡糖,并决定ABO血型。ABO位点是少数几个已经被重复验证出与多种肠道细菌丰度相关联的位点之一,包括Collinsella, Bifidobacterium以及Faecalibacterium。
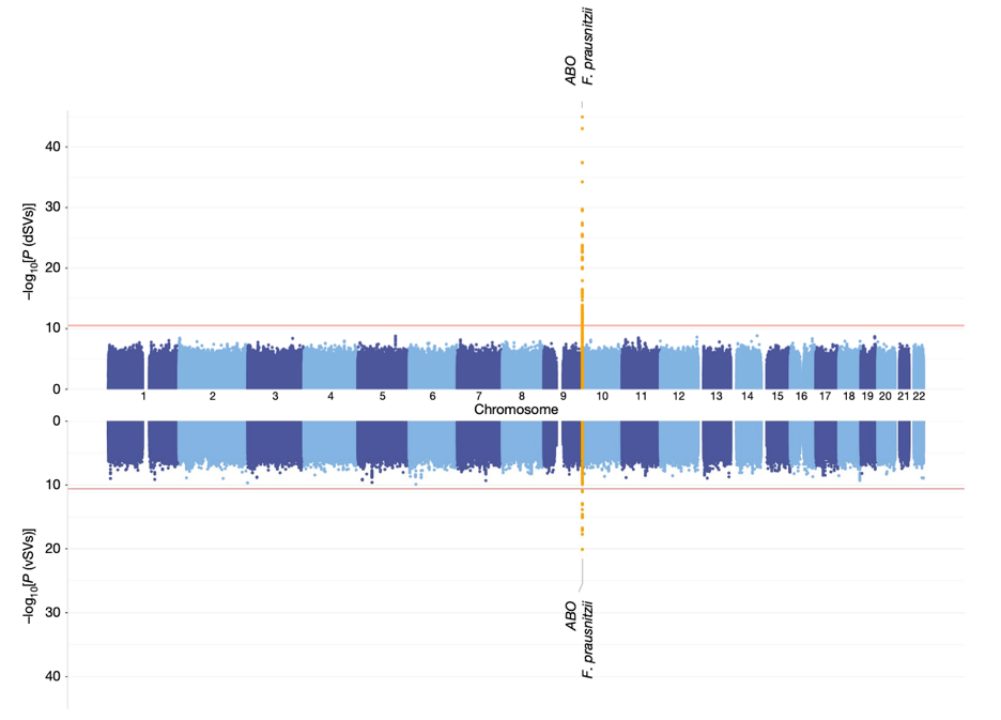
图2 人类SNP和肠道微生物SV的全基因组关联分析的曼哈顿图展示。
上方为dSV的关联分析结果,下方为vSV的关联分析结果。Bonferroni method: 3 x 10-11 for dSVs, 2.65 x 10-11 for vSV.
进一步在300TZFG队列中对所检测到的关联进行验证,该队列具有不同的遗传背景和生活方式。F. prausnitzii的SVs在该队列的201名个体中检测到。其中检测到156个与ABO基因型存在名义上显著相关的关联(p < 0.05)。其中两个F. prausnitzii dSVs (577-579bp, 575-577bp)与ABO展现出显著关联,与在荷兰队列中发现的一致(附图3c-d)。
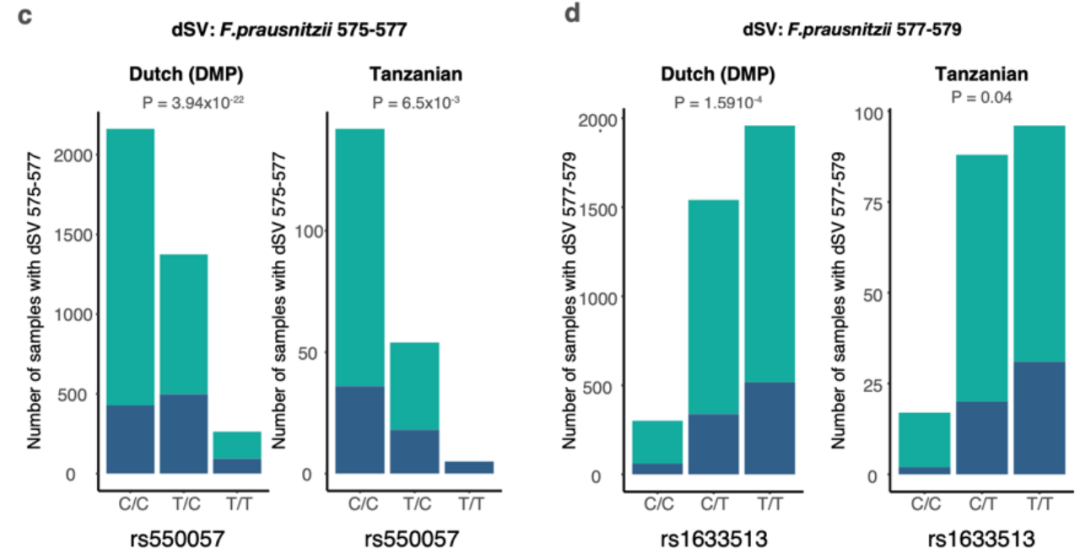
附图3c-d 荷兰和坦桑尼亚队列中F. prausnitzii dSVs 575-577与rs550057以及rs1633513的关联。
柱状条形图显示的是每个基因型具有(深蓝色)和不具有(青色)dSV区域的样本数量。
4.ABO关联依赖于FUT2基因型
由于ABO基因型决定了血型,本研究进一步在荷兰队列中分析了与ABO SNPs的关联是否能够代表与ABO编码的血型之间的关联。
结果显示,所有5个与ABO相关的F. prausnitzii SVs都与宿主的ABO血型相关。F. prausnitzii dSVs 577-579kb区域在A型或AB型血型个体中的出现频率高于B型或O型血型个体(Pmeta = 1.24 x 10-44, PDMP = 1.03 x 10-32)。
该关联还取决于FUT2分泌者的状态,该状态决定了A或B抗原的岩藻糖基前体是否分泌到体液和肠道粘液中。该分泌者决定性的SNP rs679574与dSV呈现出了暗示性的相关性(Pmeta = 2.92 x 10-9),而且仅在具有特定FUT2基因型(分秘者)的个体中,该dSV与A抗原的存在之间存在显著的关联(图3a, Pmeta = 4.85 x 10-51, PDMP_secretors = 9,39 x 10-37, PDMP_noonsecretors = 9,39 x 10-37)。在没有该基因型的个体中,这种关联并不明显。
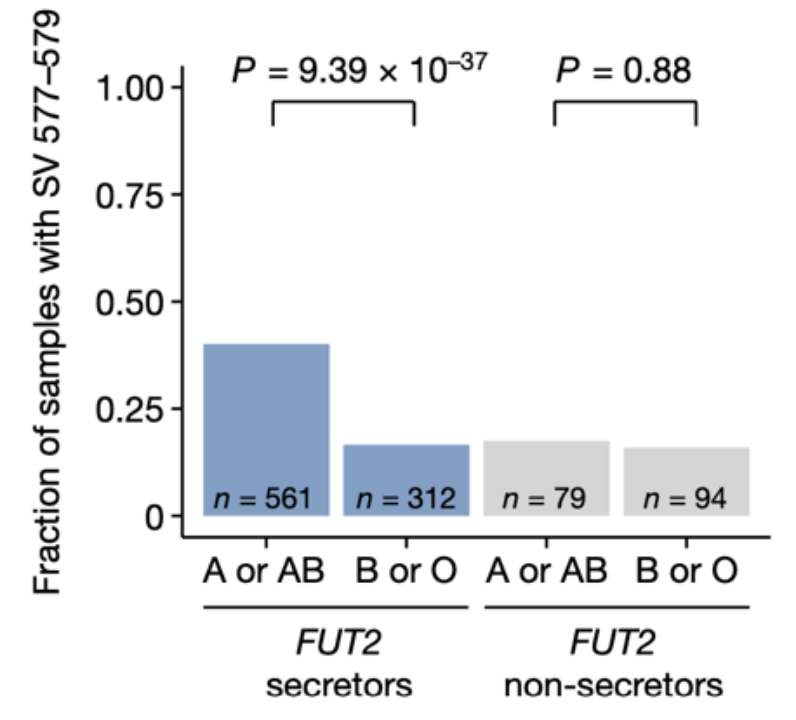
图3a ABO血型与F. prausnitzii dSVs 577-579kb区域是否存在关联取决于FUT2分秘者的状态
5. SV区域中的GalNAc代谢通路
A-抗原是一种寡聚糖,可以被分泌到肠粘膜中,并被肠道细菌的碳水化合物活性酶所降解。因此,这里本研究推断与该抗原相关联的结构变异区域可能可以赋予F. prausnitzii利用A-抗原释放的糖作为碳水化合物来源的能力。
我们发现所有的5个ABO关联的F. prausnitzii SVs很大程度上彼此之间相关联(Spearman相关系数 > 0.13, p < 0.05)。因此在将其他相关的SV作为协变量进行调整后,观察到关联强度下降,但是5个SV中的两个dSV(577-579和1154-1155)的关联在Bonferroni校正后仍然显著。其中关联性最强的dSV 577-579的结构变异区域可能捕获了大部分的信号,但不是全部。
为了进一步研究这些结构变异区域所关联的微生物基因,研究者从人类粪便中分离了F. prausnitzii, 并进行了全基因组测序,并筛选出了12种不同的F. prausnitzii菌株。结果发现,其中7个菌株显示出与ABO关联的dSV 577-579存在重叠的缺失区域,因此本研究将该2kb的dSV区域扩展到一个23-kb的区域。并以具有完整该区域(2640-2663kb)的F. prausnitzii HTF-238菌株作为参考基因组来作为基因特征的参考,从而探究位于此区域上的基因。
在该区域上共找到了27个基因,其中包括参与碳水化合物代谢的基因,特别是参与GalNAc代谢的途径,包括一组负责D-半乳糖胺和GalNAc的摄取和代谢的基因簇(图3b,c)。
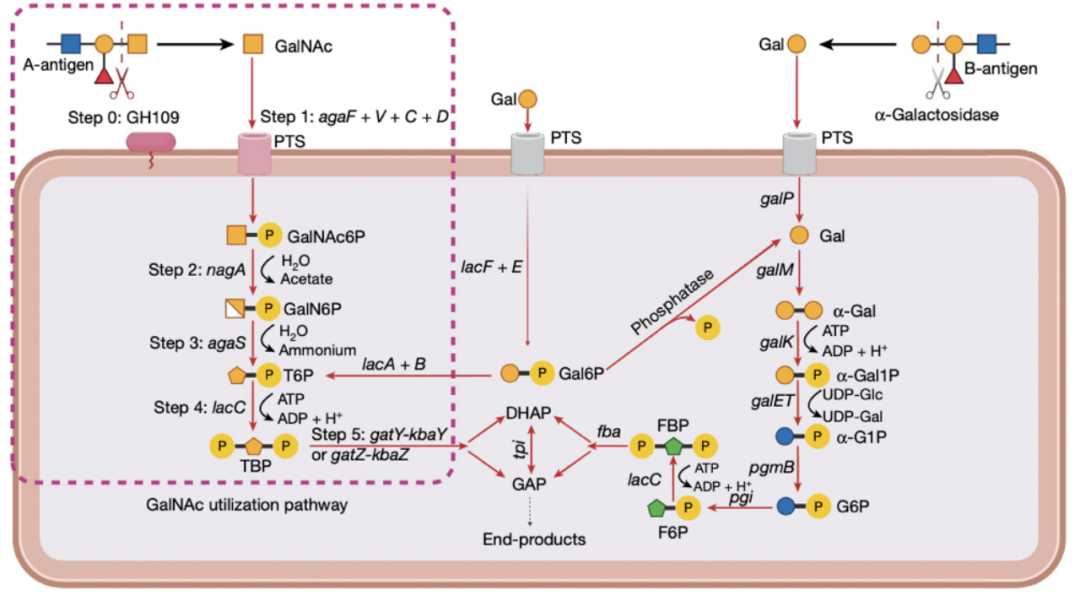
图3b 在相关SV区域中所识别到的GalNAc代谢通路。
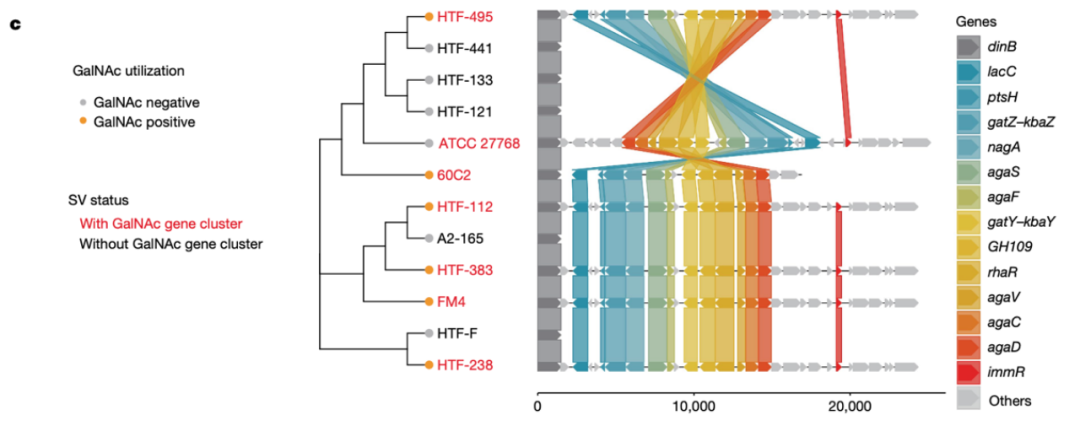
图3c 12个F. prausnitzii菌株的系统发育树。
不同颜色的线表示相同的基因在不同菌株中的位置。存在dSV区域的strain被标记为红色。
GalNAc糖是由ABO编码的A-抗原的一部分,当它被分泌到粘液中时,他可以被细菌用作能源。具体来说,该区域包含一个GH109基因,该基因编码了一种能够从A-抗原中裂解GalNAc的糖苷水解酶,以及涉及下游GalNAc利用的五个关键代谢步骤的9个基因(图3b)。此外,该区域还包含涉及半乳糖降解途径的两个基因(the Leloir and tagatose 6-phosphate (T6P) pathways)。位于该SV区域中的其他基因还包括转录调节因子、转座子以及几个未经过注释的基因,这些基因可能不直接参与碳水化合物代谢。
此外,我们发现该SV区域很可能是移动遗传元件。通过调查共同居住在一个环境里的个体之间的SV共享情况,我们发现了支持含有GalNAc的菌株在人群间传播的证据。此外,对119名个体的四年纵向分析发现,随着时间的推移,含有GalNAc的菌株的增加频率要高于减少频率(附图6)。
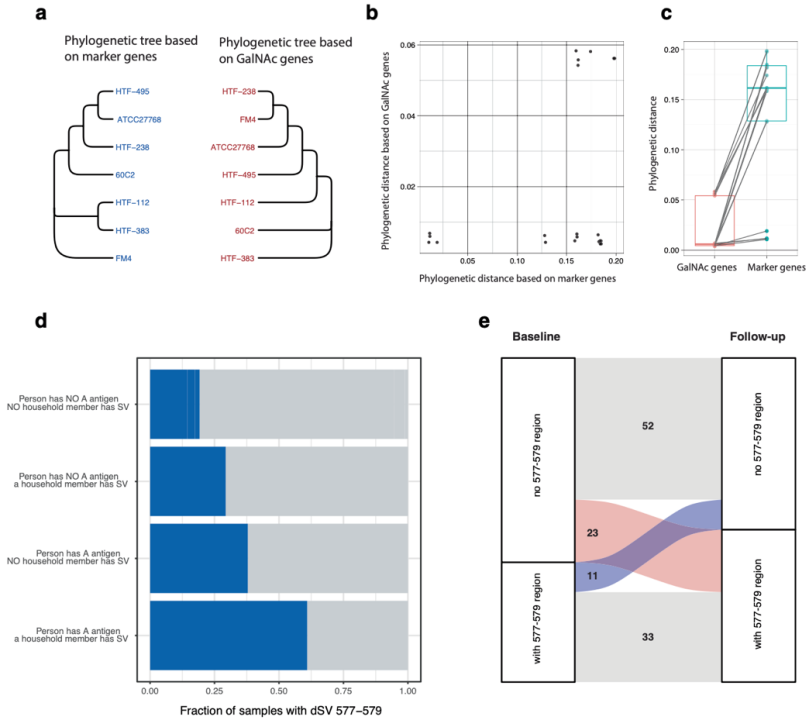
附图6 F. prausnitzii SVs的传播
a-b 基于marker基因和GalNAc基因分别绘制的系统发育树以及相应的遗传距离。
c. GalNAc基因在不同strain的遗传差异要显著小于marker基因的遗传差异,说明该基因是水平转移而获得。
d. 具有或不具有dSV 577-579的比例取决于家庭成员是否具有dSV.
e.相距四年的两个时间点的个体获得和失去dSV 577-579区域的数量。
6. 细菌可以利用GalNAc作为碳源
由于在F. prausnitzii的SV区域发现了多个参与碳水化合物的基因,我们随后调查了该区域中的基因是否对细菌利用特定单糖底物具有重要作用,包括氨基葡萄糖、半乳糖、葡萄糖、乳糖、甘露糖、N-乙酰葡萄糖胺、果糖、N-乙酰神经氨酸和2’-岩藻糖乳糖。这里选择了12株F. prausnitzii菌株进行生长速率实验,并将其培养在以上述单糖为唯一碳源的培养基(YCFA)中,将不含有碳水化合物YCFA培养基作为阴性对照。
在N-乙酰半乳糖(GalNAc)培养基中,缺乏N-乙酰半乳糖途径的菌株则无法生长,而具有N-乙酰半乳糖途径的七株菌株中的6株可以生长(图3d)。
与GalNAc利用情况相反,所有菌株都能够在半乳糖培养基上生长,但是含有Leloir和T6P途径区域的菌株比没有的菌株生长速率要更高(图3e)。这表明尽管这些途径不是必须的,但是可以提升半乳糖利用效率。并未发现该区域中途径的存在或者缺失能够影响对其他单糖的利用。
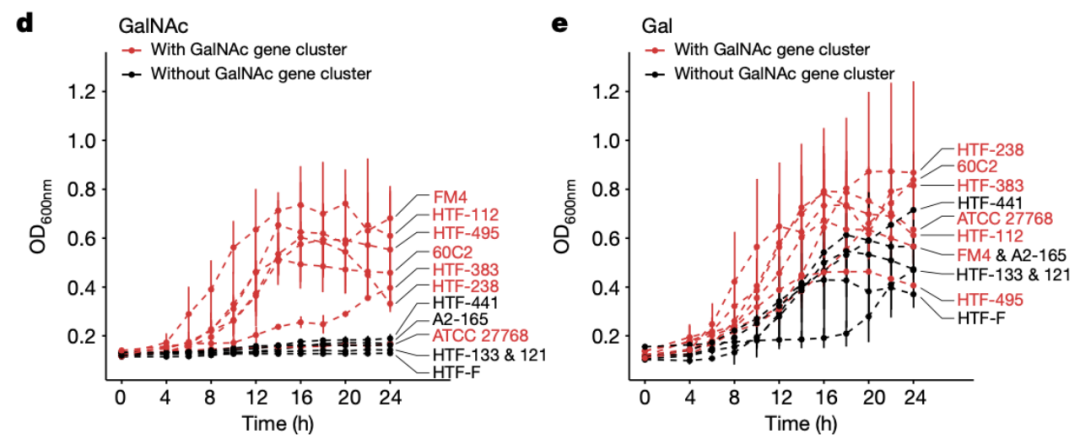
图3 d-e: 含有与不含邮dSV 577-579的菌株在含有GalNAc (d)和半乳糖(e)的培养基上的生长曲线
7. 倒位会影响GalNAc基因的表达
ATCC 27768是唯一一个携带N-乙酰半乳糖途径但却没有在N-乙酰半乳糖培养基中生长的菌株。
我们发现该菌株的N-乙酰半乳糖区域呈现倒位(图3c),可以推断出该基因的倒位可能导致了该基因功能的失调。因此,这里进一步进行了N-乙酰半乳糖诱导实验,以研究该区域内N-乙酰半乳糖基因和潜在调控因子(ptsH, rhaR和immR)的转录。
N-乙酰半乳糖诱导实验:将ATCC 27768在葡萄糖培养基中进行预培养,随后将产生的细菌培养分为两个部分,分别转移到半乳糖培养基中和N-乙酰半乳糖培养基中。然后比较了两个培养基中基因表达倍数变化。阳性对照是近缘菌株HTF-495,他可以在N-乙酰半乳糖培养基中生长,而阴性对照则设置为缺乏N-乙酰半乳糖基因簇的HTF-441。
研究结果显示,在HTF-441红未检测到N-乙酰半乳糖基因的表达,证实了它们的缺失。值得注意的是,在N-乙酰半乳糖诱导之后,ATCC 27768中是三个N-乙酰半乳糖摄取基因agaC, agaD和agaV的表达量仅仅略有增加,而这些基因在HTF-495菌株中则表现出明显增加。例如,与葡萄糖诱导相比,基于N-乙酰半乳糖诱导的HTF-495菌株中agaC基因的表达量增加了63.5倍,而在ATCC 27768中仅增加了三倍(图3f)。因此这些结果表明,ATCC 27768的基因逆转仅影响了N-乙酰半乳糖摄取基因的表达,而不影响N-乙酰半乳糖代谢基因。
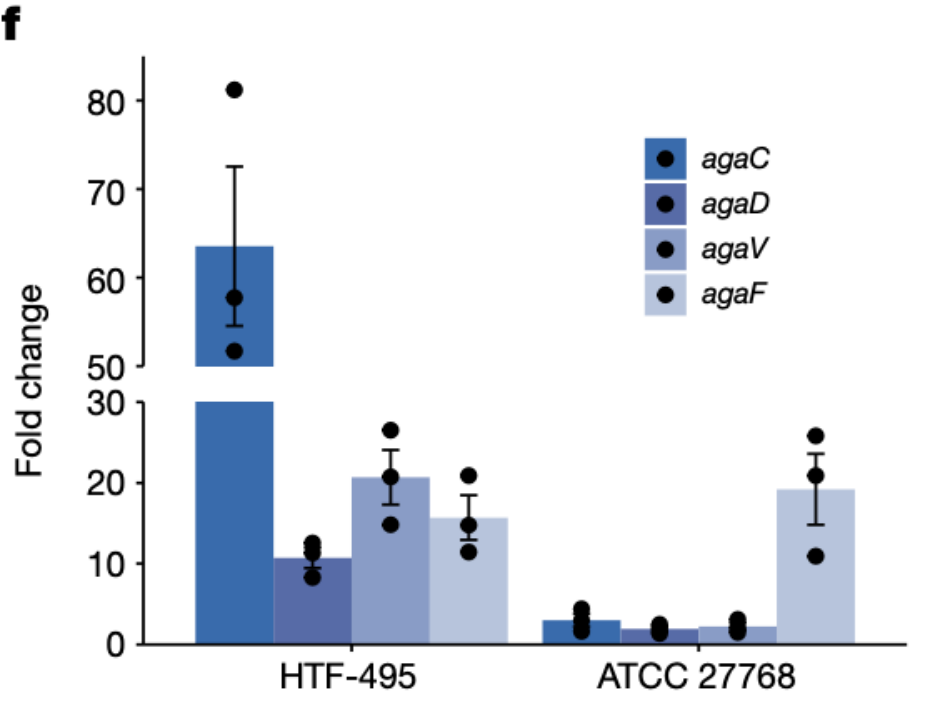
图3f 与葡萄糖诱导相比,GalNAc诱导后的基因表达的倍数变化。
8. 其他细菌物种中的GalNAc代谢途径
迄今为止,之前的研究已经发现ABO位点与9个细菌物种的丰度相关联,其中包括三个物种:C. aerofaciens、Faecalicatena lactari、Bifidobacterium bifidum. 然而,仅有Collinsella属的关联能在其他研究中得到验证。因此,这里本研究想要知道GalNAc途径的存在与否是否可以解释ABO位点与这些细菌物种丰度的关联。
这里,本研究从HUGG数据库中提取了10467个ABO相关微生物的组装基因组:1103个C. aerofaciens, 484个F. lactari,1109个B. bifidum 以及7791个F. prausnitzii. 然后对这些物种进行了GalNAc基因的同源基因搜索,我们发现28%-95%的组装基因组中存在GalNAc基因(图4a)。然而只有2678个组装基因组中具有完整的途径,其中包括1784个F. prausnitzii菌株(23%)和884个C. aerofaciens菌株(80%)(图4b-c)。因此C. aerofaciens菌株中高比例的携带GalNAc可能可以解释Collinsella属与ABO位点之间的关联。
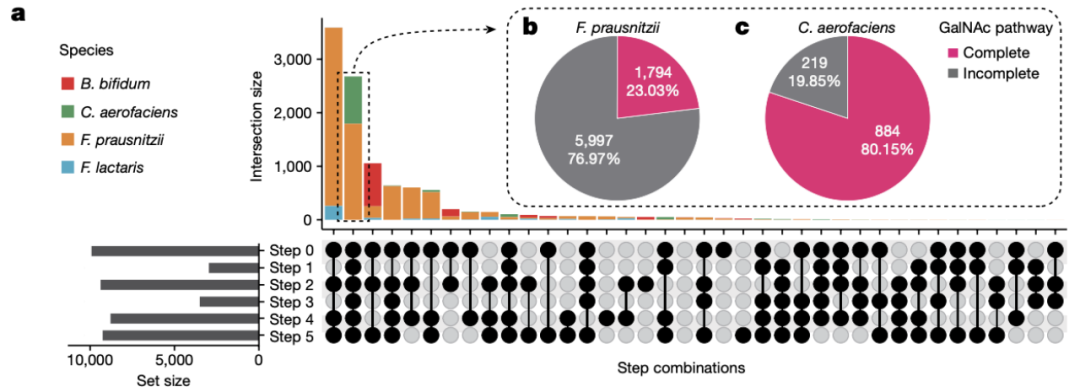
图4 F. prausnitzii与其他相关物种的GalNAc的利用能力。
a.与人类ABO血型相关的四种肠道微生物物种中拥有完整GalNAc的数量。
b-c: F. prausnitzii和C. aerofaciens中具有完整GalNAc途径的菌株比例。
此外,我们还在两个C. aerofaciens菌株中证明了其GalNAc的利用能力(图4d-g)。
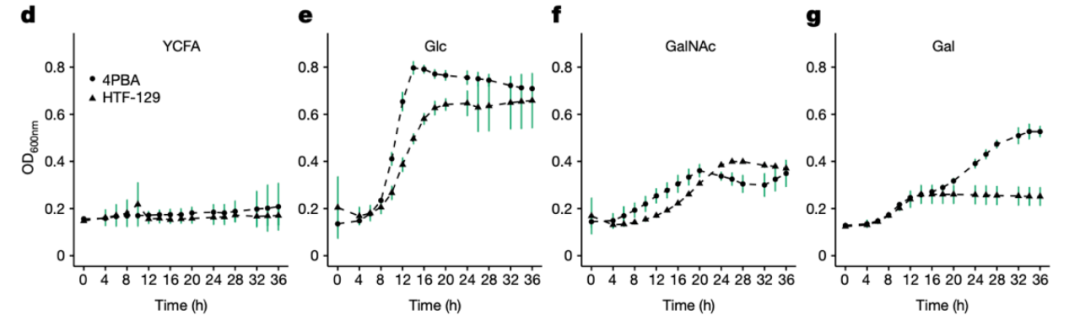
图4d-g 携带GalNAc通路的两个C. aerofaciens菌株在含有不同糖的培养基上的生长曲线。
然而在B. bifidum菌株中并未找到完整的GalNAc途径,这表明B. bifidum与人类血型关联的潜在不同机制。
9. GalNAc的利用对于人类健康具有支持作用
本研究进一步估计了整个微生物群落中GalNAc基因簇的总丰度。这些GalNAc基因表现出强烈的互相相关的性质,这表明这些基因通常以基因簇的形式存在,并且共同发挥着功能。
同样的,本研究也发现GalNAc途径中关键基因的表达水平与FUT2分泌者的ABO血型相关联(附图9)。而且在基因水平上观察到的关联要比SV水平上观察到的关联显著的多,p值可以达到4.19 x 10-223.
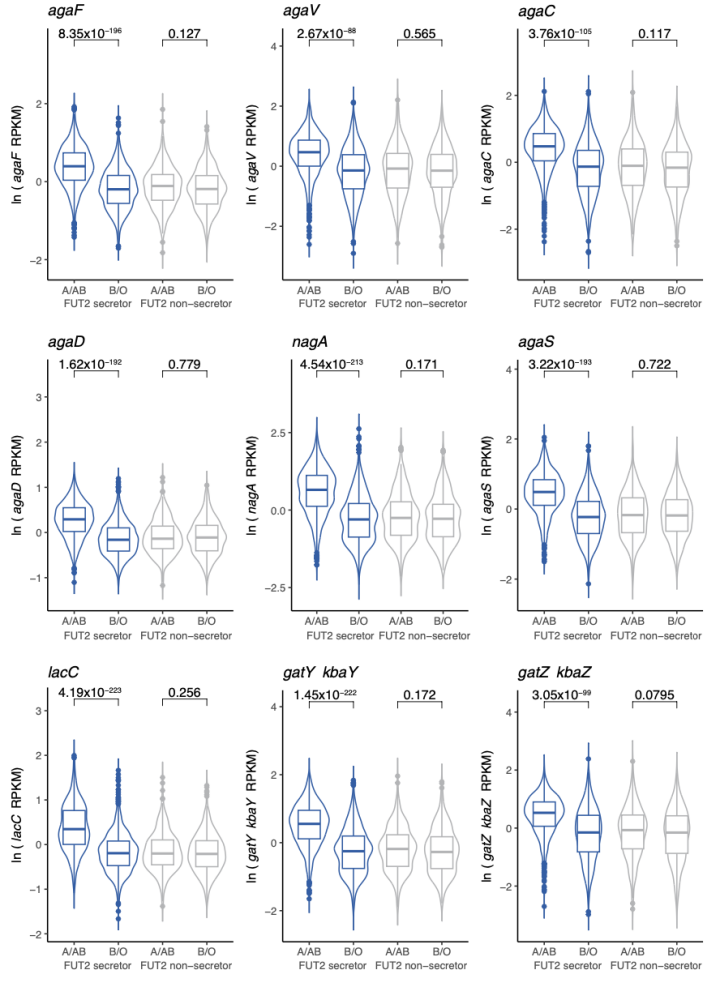
附图9 9个关键GalNAc通路基因与FUT2分泌者和非分秘者中A抗原存在的关联。
这里进一步推断GalNAc基因的表达水平对于具有黏膜A抗原的个体的健康更为关联,而对于没有黏膜A抗原的个体则不太相关。
因此,为了验证这一假设,这里根据ABO和FUT2基因型将队列中个体分类为在长达粘液中具有和不具有黏膜A抗原的个体。其中具有A抗原(A型或AB型)的FUT2分秘者被定为师具有黏膜A抗原的个体,其他所有人则被认为是不具有黏膜A抗原的个体。
与之前的研究一致,具有和不具有黏膜A抗原的个体在GalNAc的基因表达水平上存在显著差异。其中两个基因与GalNAc代谢途径中的关键催化步骤之间的关联:即基因lacC,其参与了T6P (tagatose 6-phosphate)到tagatose 1,6-二磷酸的第四个催化步骤(P = 1.30 x 10-280)。第二个基因是gatY-kbaY,它参与了从tagatose 1,6-二磷酸到二羟基丙酮磷酸或甘油醛-3-磷酸的第5个催化步骤(P = 2.60 x 10-259)(图5a,b)。
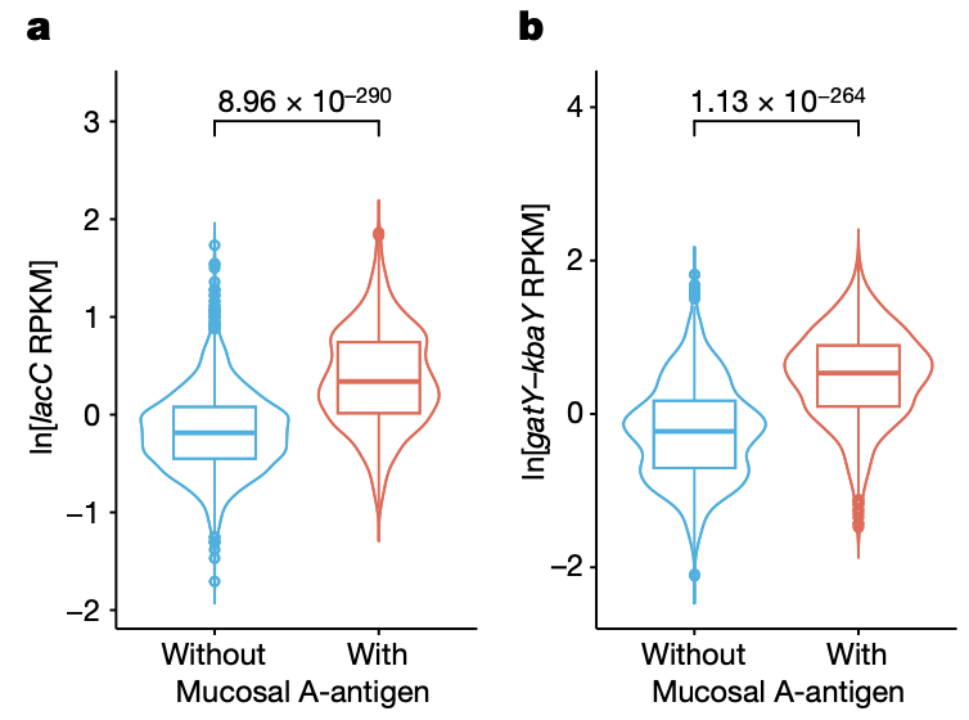
图5 有无黏膜A抗原的两组个体之间GalNAc关联的比较。
(a-b)两组个体之间GalNAc基因 lacC (a)和gatY-kbaY (b)表达水平的比较。
由于许多肠道微生物都可能具有GalNAc途径,我们这里进一步推断,黏膜A抗原的存在可能提供额外的能源来源,以促进GalNAc利用者的生长。与该假设一致,本研究发现GalNAc基因的丰度与微生物的丰富度和多样性呈现正相关,并且这种关联在具有黏膜A抗原的个体中要更为显著(Pheterogeneity < 0.05, I2>0.7,图5c)。
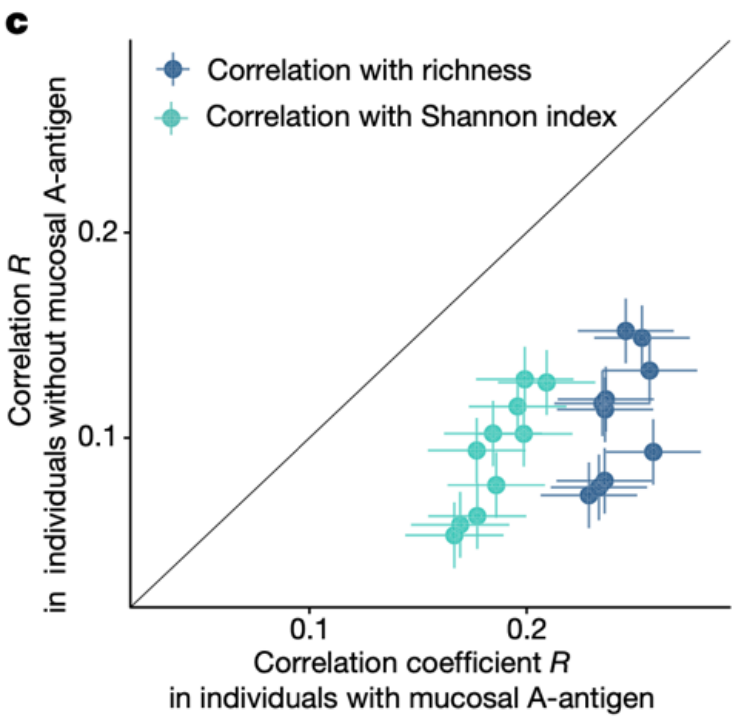
图5c 比较两组个体GalNAc途径丰度与肠道微生物多样性和丰富度相关性,每个点代表GalNAc与香农指数或者丰富度之间的Spearman相关系数。
同样的,我们将微生物GalNAc基因与240个环境包括和健康相关的参数在具有和不具有黏膜A抗原的个体中进行关联。在经过Bonferroni P值校正后,在存在A抗原的组中检测到了50个显著关联,而在A-抗原缺失的组中只检测到了17个关联。值得注意的是,GalNAc基因的丰度与血糖、Bristol类型以及GeneralHealth仅在具有黏膜A抗原的组中显著相关(图5d,e)。尽管有的关联在不具有A抗原的组中也检测到,但是关联程度相比另一组较小。
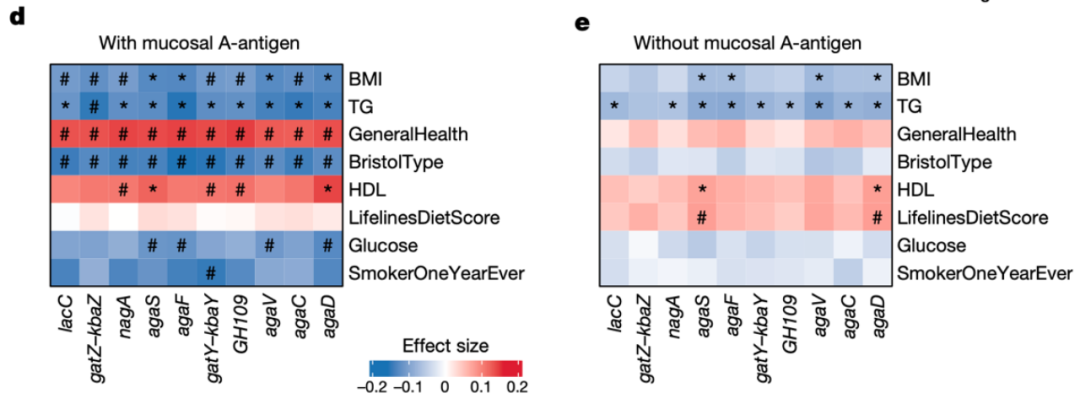
图5d,e 具有和不具有黏膜A抗原的个体中GalNAc代谢基因丰度与宿主表型的关联。
总结
对来自四个荷兰队列的9015个个体进行了宿主遗传变异和肠道微生物结构变异之间进行了全基因组关联研究。我们发现,人类ABO编码的A血型与F. prausnitzii中一个含有GalNAc代谢基因簇的基因组片段存在强相关。而且这一关联在坦桑尼亚队列中得到了复制。菌株培养实验表明,GalNAc途径对于将GalNAc用作碳水化合物来源至关重要,这一结果也解释了之前研究所观察到的ABO位点与F. prausnitzii和C. aerofaciens相对丰度之间的关联。
之前的研究通常是将微生物的丰富度与宿主遗传联系起来,然而通常发现遗传效应对于微生物丰度的影响较小。尽管已经尝试将物种丰富度扩展到功能水平上,但是这些分析通常是基于代谢途径的注释,而注释程度远未完善。这里我们的研究表明,将宿主遗传和细菌SV关联起来有助于确定可能的治病基因,从而可以弥补从物种丰度到功能的差距。而且,在本研究中,我们将物种的丰度作为协变量,以识别独立于物种丰度的特定SV区域的关联。因此研究结果会突显了从丰度转向功能途径和基因水平的重要性,从而可以更好的理解宿主遗传对肠道微生物的影响。在本研究中也证明了这一点。即ABO位点中,由FUT2分泌者ABO基因型编码的A或AB血型与细菌GalNAc基因丰度相关联,并与F. prausnitzii中含有GalNAc途径的SV区域相关,而该队列中并未发现ABO血型与F. prausnitzii丰度的关联。
在遗传上倾向于分泌黏膜A抗原的个体中存在GalNAc基因可能有益于人类健康,此外,GalNAc基因可以通过基因倒位变得功能失调,并且它们可以在细菌之间传播并在人类之间共享。
ABO血型与人类健康之间存在广泛关联,包括静脉血栓栓塞,血脂水平以及其他心脏代谢表型,以及对多种传染病的易感性和严重程度。因此ABO基因座与人类健康中的广泛重要性突显了本研究中基于人类微生物组关联的重要性。ABO与细菌GalNAc代谢基因之间的墙关联,以及后者与微生物多样性和丰富度的关联,支持了一个新的假设,即ABO可能通过对肠道微生物组的影响来影响人类健康。因此,增加GalNAc利用菌株例如F. prausnitzii和C. aerofaciens可能有益于增加微生物多样性。与此一致,本研究还表明,细菌GalNAc基因丰度在存在黏膜A抗原的个体中与人类健康呈现正相关。
相关数据
识别到的结构变异概况以及,细菌dSV和vSV相关的遗传关联统计数据:https://doi.org/10.25452/figshare.plus.c.6877849.
关于简说基因
生信平台
Galaxy中国(UseGalaxy.cn)致力于打造中国人的云上生物信息基础设施。大量在线工具免费使用。无需安装,用完即走。活跃的用户社区,随时交流使用心得。
生信分析
我们能够承接所有 NGS 组学数据分析业务,包括但不限于 WGS / WES / RNA-seq 等。基因组组装、注释,以及各种重测序业务都可以与简说基因合作。
生信培训
简说基因的生信培训班,荣获学员的一致好评。如果你也对生物信息学感兴趣,欢迎来跟简说基因,学真生信。
联系方式
QQ交流群(免费):925694514
微信交流群(免费):加微信好友,注明“Galaxy交流群”
客服微信:usegalaxy














 7237
7237











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









