






大麦是全球重要的农作物,其遗传多样性对农业生产至关重要。近年来,基因组学技术的发展为大麦的遗传研究提供了新的机遇,泛基因组的研究有助于深入理解大麦的遗传变异。今天我们就一起来学习下面这篇关于“剖析大麦泛转录组,洞察遗传与转录互作机制”文章的研究思路和生信分析方法。

论文链接:
https://doi.org/10.1038/s41588-024-02069-y
大麦泛转录组数据库(PanBaRT20):
https://ics.hutton.ac.uk/panbart20/index.html
该研究由英国皇家生物学会院士Robbie Waugh领衔,张润烜博士、过文彬博士团队联合英国詹姆斯·赫顿研究所及多家国际研究机构共同完成。研究旨在构建大麦泛转录组,揭示基因型与转录组之间的关系,为大麦的遗传育种和功能基因组学研究提供重要的基础数据,数据分析部分用到了Fastp、STAR、Stringtie、Salmon、InterProScan等一系列生信工具,大部分工具在Galaxy生信云平台(网址:usegalaxy.cn )都有部署,非商业纯学术用途均可免费在线运行,无需编程无需安装工具和配置复杂的环境。
研究结果
泛转录组构建
• 数据来源与处理:从 20 种大麦基因型的五个组织中获取 RNA 测序和 PacBio Iso - seq 数据,经过严格的质量控制和筛选,确保数据的准确性和可靠性。
• 转录本多样性:
基因数量与转录本数量:构建的20个基因型特异性参考转录数据集(GsRTDs),GsRTDs 包含的基因数量在 35,500 到 40,800 之间,平均每个基因有 3.22 个转录本,显著高于传统基因组注释。
不同品种对比:Barke GsRTD 与近期发表的参考转录数据集相似,表明 GsRTDs 的高质量(图 1),表明构建的转录数据集具有可靠性和可参考性。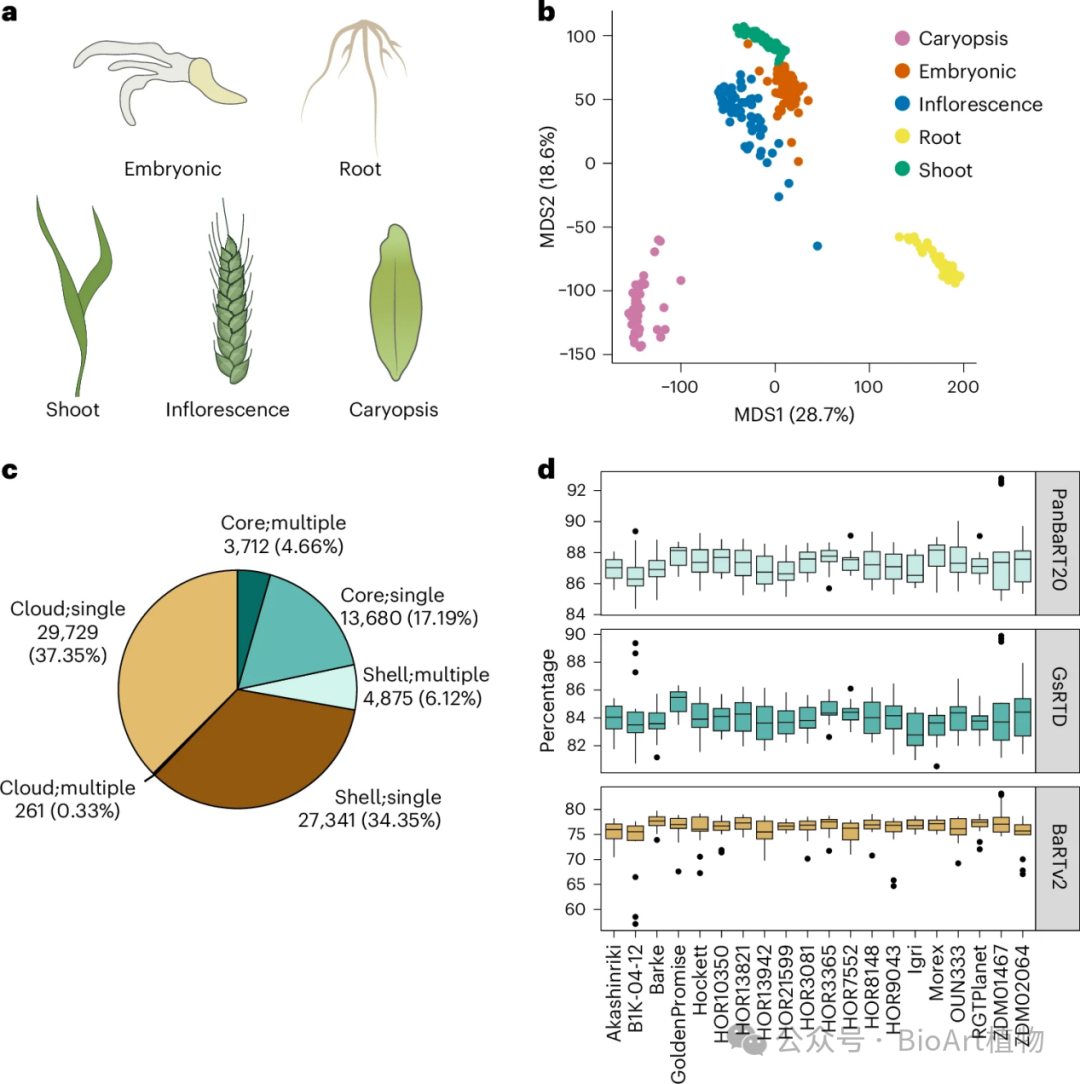
图1:转录本多样性和分类
• 基因分类:
分类标准与比例:依据基因在不同基因型中的表达情况,将基因分为核心、外壳和云状三类,占比分别为17.19%、4.66%和37.69% 。这种分类方式为研究不同基因的功能和特性提供了重要依据。
功能关联:GO 富集分析表明,核心基因主要与普遍存在的生物学功能相关,外壳和云状基因主要参与生物和非生物胁迫响应,比如应对病虫害侵袭、适应环境变化等(图1c、扩展数据图2b)。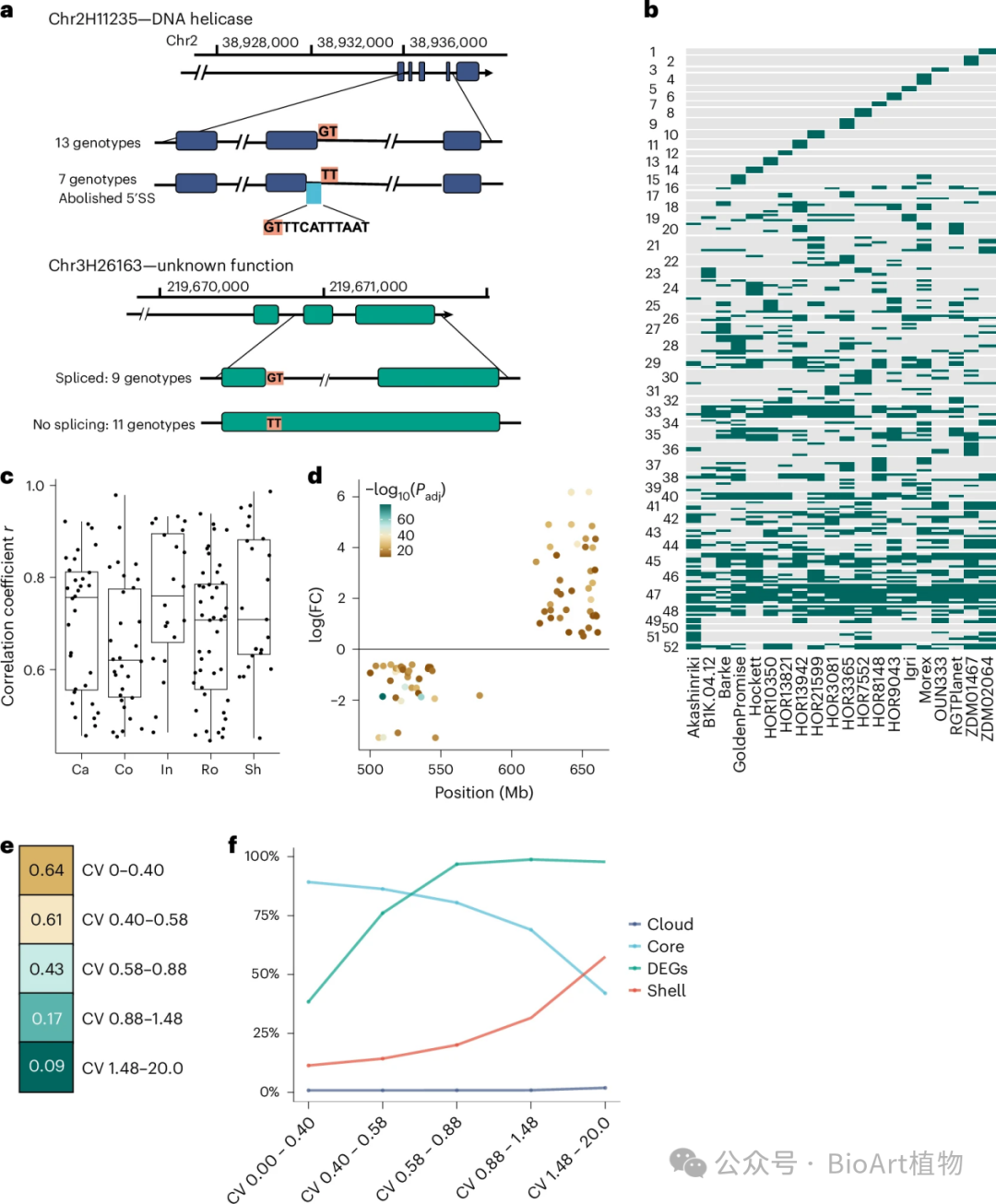
图2:转录本丰度变化的驱动因素。
转录本丰度变化
• 整体特征
转录本数量增加:PanBaRT20中基因的转录本数量从GsRTDs的平均3.5个显著增加到7.3个(图2a),反映出在泛转录组层面基因表达的复杂性和多样性进一步提升。
剪接变异增加:非冗余剪接接头数量从GsRTDs的平均146,600个增加到PanBaRT20的311,300个,表明基因在转录过程中的剪接方式更加多样化,丰富了转录本的种类。• 影响因素
基因变异:像chr2H11235和chr3H26163等基因发生突变,导致剪接位点改变,进而产生独特的基因型特异性转录本(图2a),展示了基因变异对转录本的直接影响。
拷贝数变异:CBF2和CBF4等基因的拷贝数与转录本丰度紧密相关,高拷贝数往往会导致更高的基础基因表达(图2c、扩展数据图3a),体现了基因剂量对转录的调控作用。
染色体结构变异:染色体7H上的倒位使得倒位区域内基因表达差异显著,并与多种性状相关联,如影响作物的株高、产量等(图2d、扩展数据图3b - d)。
共表达网络分析
• 网络构建:通过 WGCNA 构建了 20 个基因型特异性的共表达网络,共发现 738 个模块(图3a),为研究基因之间的协同表达关系提供了直观的网络模型。
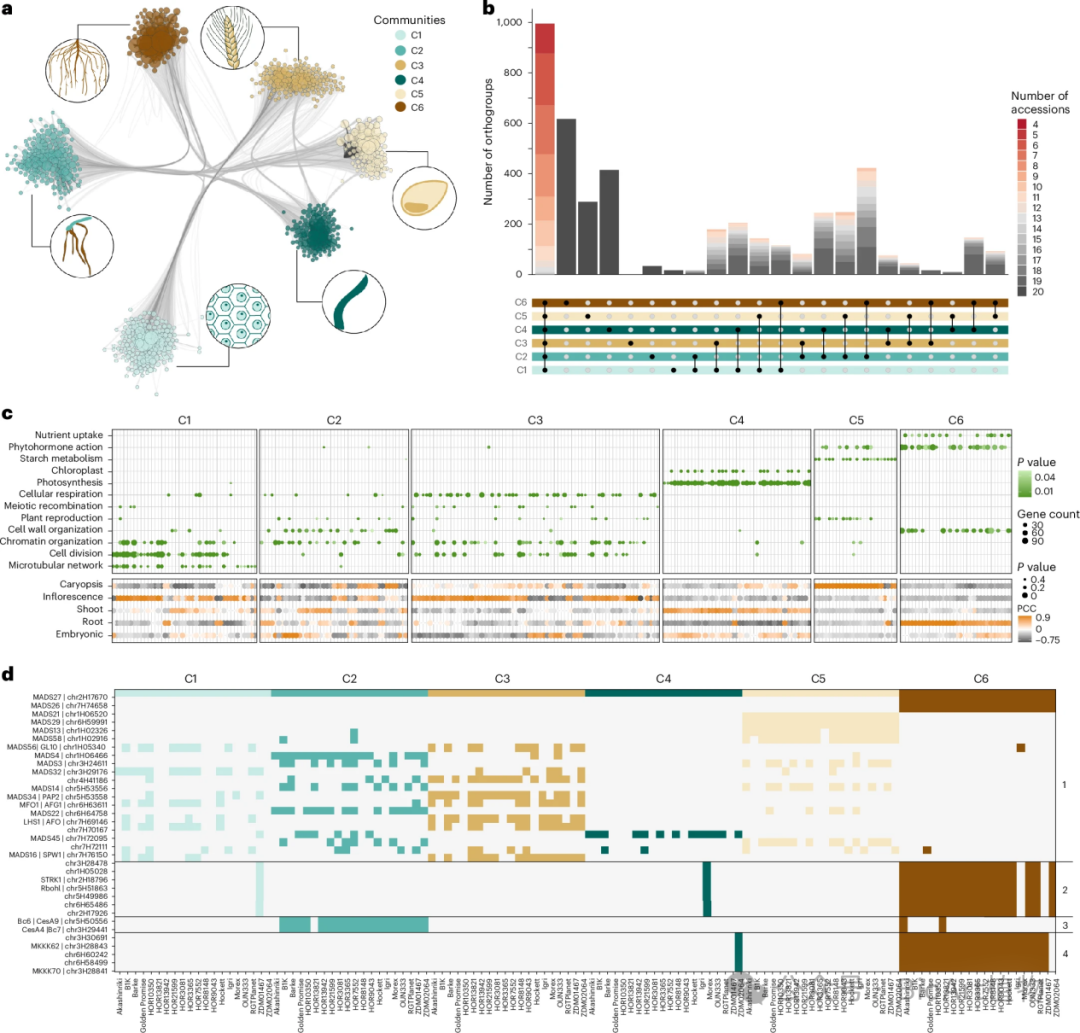
图3:比较基因表达和基因网络分析。
• 模块特征
模块内基因相关性高:同一模块内的基因表达具有较高的相关性,如模块 C4、C5 和 C6 与特定组织和生物学过程显著相关,它们可能共同参与某一代谢途径或生理过程。
模块间差异明显:不同模块之间的基因表达模式差异显著,反映了基因在不同组织和生物学过程中的特异性表达,暗示了基因功能的特异性和分化。• 基因分布与功能
同源基因多样化:同源基因在共表达网络中呈现出功能多样化,以Mkkk62和Mkkk70为例,它们在不同基因型中的表达存在差异,表明同源基因在不同遗传背景下功能有所分化。
转录因子作用:转录因子如 MADS - box 蛋白等在网络中整合了许多发育信号,对基因表达的调控具有重要作用。
Morex基因表达图谱
• 表达特征:Morex 基因表达图谱揭示了该品种在不同组织和处理条件下的基因表达模式,发现了一些在 PanBaRT20 中未检测到的基因表达差异(图4)。
• 新基因发现:通过与 PanBaRT20 比较,发现了 5,230 个在 PanBaRT20 中没有序列匹配的基因,这些基因可能是特定组织和条件下的新基因,为大麦基因研究开拓了新方向。
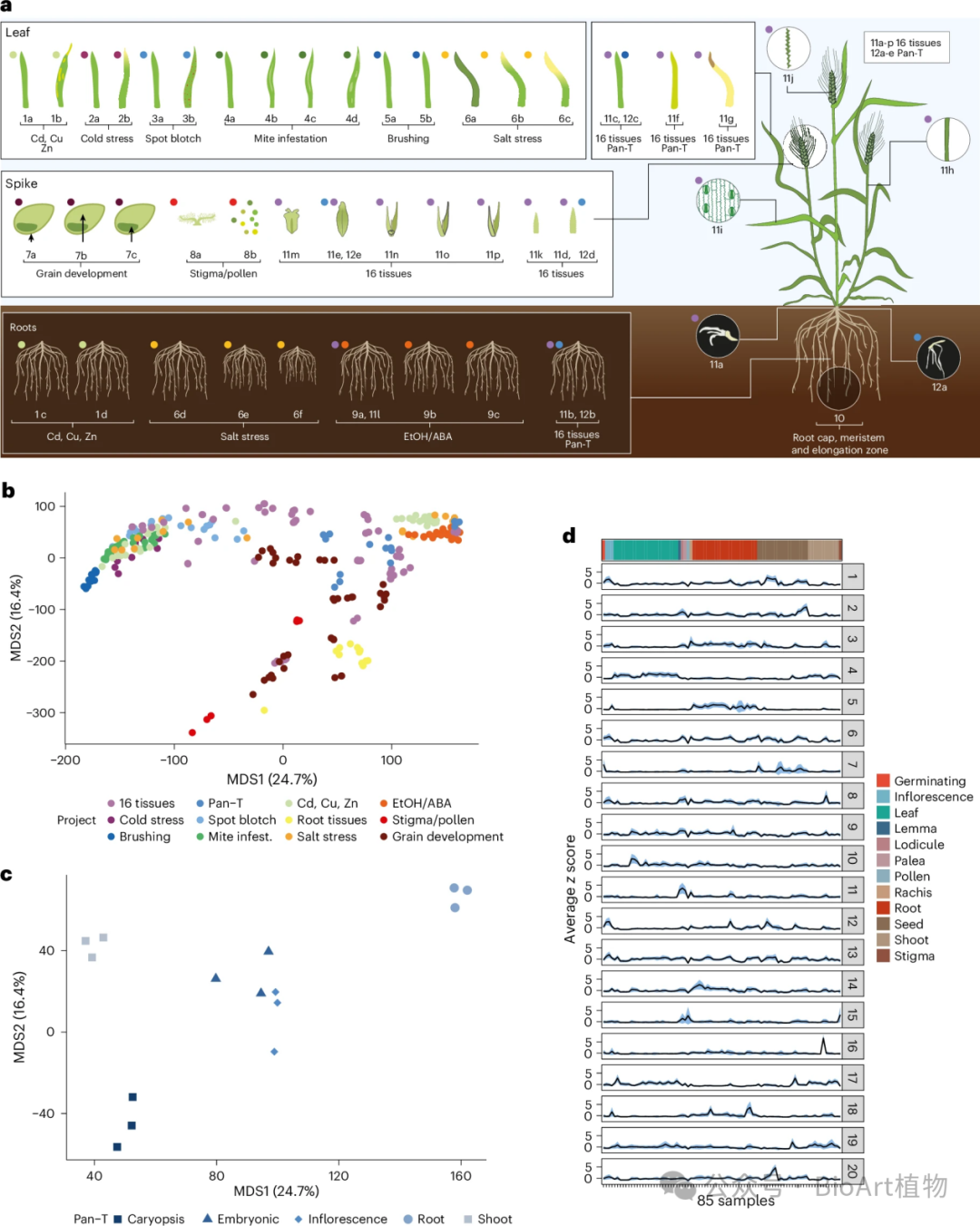
图4:Morex Atlas RNA-seq数据集和样本间的变异性。
GA2ox基因家族分析
• 基因表达:GA2ox7 和 GA2ox3 在不同组织和基因型中的表达存在差异,与植物激素信号通路相关,参与植物激素的合成与代谢调控,影响植物的生长发育进程。
• 突变体影响:ga2ox7 突变体导致产量、千粒重和淀粉含量降低,ga2ox3 突变体在特定条件下农艺性状下降,明确了这两个基因在大麦生长发育和产量形成中的关键作用。
研究方法
植物材料与处理
• 材料选择:选用 20 种大麦基因型,代表了大麦的遗传多样性,确保研究结果的代表性。
• 培养条件:对种子进行发芽和培养,控制光照、温度和湿度等条件,确保植物生长在相同的环境中。
RNA提取与测序
• 提取方法:使用多种试剂盒提取总 RNA,包括 Macherey - Nagel NucleoSpin RNA Plant Mini Kit 和 Invitrogen 的植物分离辅助试剂,确保 RNA 的质量。
• 测序技术
Illumina测序:进行短读长 RNA 测序,生成 150 bp 的配对末端测序数据,用于基因表达的定量分析。
PacBio Iso - seq测序:进行长读长测序,用于转录本的全长测序和组装,提高转录本的准确性和完整性。
数据分析
• 转录本组装
软件工具与策略:使用 Fastp、STAR、Stringtie 等软件对 RNA - seq 和 Iso - seq 数据进行处理和组装,采用两步法映射读取,提高转录本的组装准确性。
质量控制:使用Fastp软件去除质量分数低于20且长度小于30个碱基的读段;用STAR软件将修剪后的读段映射到参考基因组;使用Stringtie和Scallop软件构建转录本组装;利用RTDmaker软件合并组装结果,并过滤掉具有非规范剪接位点、支持读段数少于规定阈值、片段长度不足等特征的低质量转录本。
• 泛转录组构建
泛基因组构建:利用PSVCP构建以Morex V3为参考的线性泛基因组,整合20个参考大麦泛基因组,通过多轮比对、校正等操作,确保基因组的完整性和准确性。
转录本映射与分组:将 20 个 GsRTDs 的同源转录本映射到泛基因组上,根据基因结构、剪接位点等特征将转录本分组,定义核心、外壳和云状基因。• 转录组定量与分析
定量方法使用 Salmon 进行转录本定量,3D RNA - seq 进行差异表达分析,确保定量结果的准确性和可靠性。
数据分析工具
基因功能注释:使用 InterProScan 等工具对基因进行功能注释,了解基因的生物学功能。
共表达网络分析:运用WGCNA等软件构建共表达网络,分析基因之间的表达关系和功能模块。首先使用R包DESeq2进行方差稳定化变换以进行归一化,应用“~Tissue”设计并设置“blind = FALSE”;然后使用WGCNA包构建共表达网络,自动确定软功率阈值,计算Pearson相关系数以获得邻接矩阵;再使用动态树切割和TOMtype方法进行模块划分,合并紧密聚类的模块;最后使用Netgraph包计算最终的网络布局。
转录因子结合位点分析:使用FIRE等算法识别转录因子结合位点,探讨基因表达的调控机制。具体过程包括选择高质量的CDS序列,通过BLAST Best Reciprocal Hits方法确定基因对应关系;对每个基因的上游2 kb序列进行分析,扫描30个转录因子结合位点;通过SeqPattern和OmicsBox软件获取相关统计信息,计算表达一致性并进行基因分类和功能富集分析。
Galaxy云平台实现零代码生信分析
Galaxy生信云平台(网址:usegalaxy.cn )是一个在线生物信息学分析平台,提供1300多种工具,支持从数据上传、分析到结果可视化的全流程操作,适用于学生、教师和科研人员。这篇文章的生信分析用到了之前我们介绍过的 Fastp、DESeq2、STAR、Stringtie、 Salmon、 InterProScan等工具,这些工具都能在Galaxy生信云平台上方便地使用,不需要安装软件和配置复杂的环境。对这些工具感兴趣的读者可以参考(点击下方蓝色字体跳转阅读)以下文章:
DESeq2:生信零基础也能做转录组差异表达分析
InterProScan:崩溃,做生信10年,发现好累
推荐阅读
中国银河生信云平台(UseGalaxy.cn)以“让生信分析更简单”为使命。平台致力于为科研工作者、医疗机构和生物产业技术人员提供全栈式生物信息学分析解决方案。
优先技术响应、定制化工具部署、阶梯式能力培养,请加入「Galaxy生信星球」。咨询微信:usegalaxy 或 galaxy-help



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









