






TidyMass: 一个面向对象的LC-MS数据可重复分析计算框架
简介
Nature Communications于7月27日刊发了来自斯坦福大学遗传系的最新工作: TidyMass: an object-oriented reproducible analysis framework for LC–MS data.
可重复性、可追溯性和透明度(Reproducibility, traceability, and transparency)一直是LC-MS数据分析的长期存在的问题。虽然现在已经开发了多种工具,但是他们仍然存在着局限性。因此,我们开发了 tidyMass项目(https://www.tidymass.org/),tidyMass是一个基于R语言的综合计算框架,可以实现LC-MS数据的可追溯、可共享和可重复的数据处理和分析工作流程。TidyMass是一个共享底层设计理念、语法和数据结构的R包生态系统,它提供了一个全面的、可重现的和面向对象的计算框架。模块化架构使tidyMass成为一个高度灵活和可扩展的工具,其他用户可以改进并与其他工具集成以定制自己的管道。
tidyMass现在主要支持LC-MS的非靶向代谢组学的数据分析,也支持一部分的蛋白质组学的数据分析.后续开发者将继续完善其用于蛋白质组学的全流程分析.
本文共同第一作者为申小涛博士(斯坦福大学),闫红博士(耶鲁大学),和王楚楚博士(斯坦福大学),通讯作者为斯坦福大学的Michael Snyder教授,和耶鲁大学的Carline Johnson教授.参与者还包括匹兹堡大学的高鹏教授.
文章链接
https://www.nature.com/articles/s41467-022-32155-w.pdf

使用
欢迎大家使用tidyMass.我们提供了详细的tutorials,可以在 tidyMass官网找到: https://www.tidymass.org/.也可以点击阅读原文获得.

tidyMass设计理念
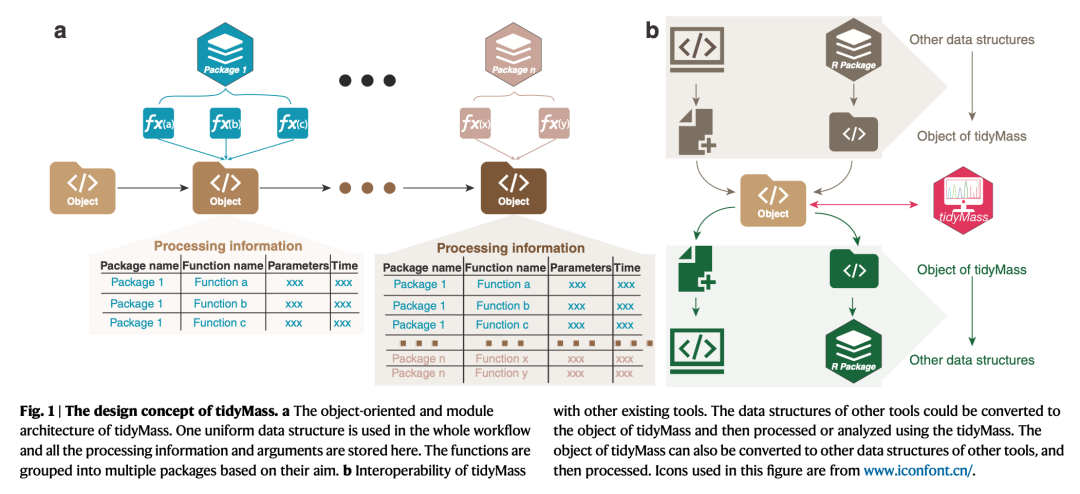
借鉴R语言中非常流行和强大的tidyverse设计理念,tidyMass首先是一个模块化设计的项目(modular architecture).tidyMass不是一个包,而是一系列包的集合.每一个包都包含了相同功能的函数.比如,所有用于代谢物鉴定的函数都集合为了metID package(https://metid.tidymass.org/).模块化设计带来的好处是,灵活性和拓展性,这在现在的R包设计中非常流行. 除此之外,tidyMass是一个面向对象的工作流,它使工作流更简单、更易于使用。统一的数据结构(对象)是tidyMass的内核,专门用于存储和管理处理后的代谢组学数据。所有功能在整个工作流程中采用统一的数据结构,与其他工具相比,这有利于用户准备输入的数据,此步骤所需的时间更少。最后一个概念是tidyMass与其他现有工具具有很高的互操作性,这是一个关键特性(因为领域内已经开发了许多强大的工具)。为了避免重复造轮子,tidyMass使用了统一的数据结构,该数据结构可以与其他工具的数据结构进行转换,从而实现与其他工具的集成使用。
统一的数据结构“mass_dataset”
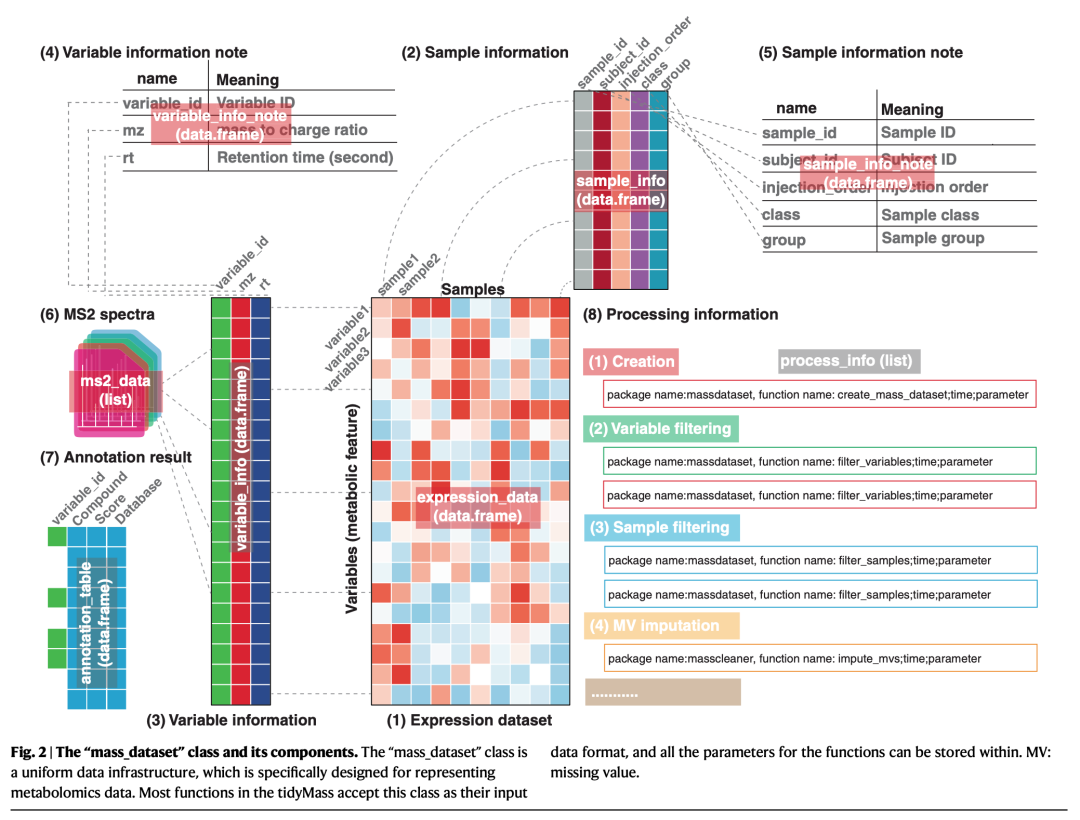
我们首先设计了一个统一数据结构(“mass_dataset” class)来存储和管理非靶向代谢组学数据, 包括表达量数据、样品和变量信息(metadata)。此外,数据的内容是自动同步的,当用户操作一个内容时,它将自动将操作传播到所有相应的内容。这使得操作和维护数据的一致性变得容易。tidyMass中的所有函数都使用“mass_dataset”作为它们的输入数据格式,因此一个数据结构可以用于所有处理和分析步骤。此外,“mass_dataset”类支持来自其他包的流行工具,特别是tidyverse,它是R语言中数据科学使用最广泛的工具之一。这样的设计让tidyMass的代码更加通用和直接,这对新用户来说是有利的,他们不需要学习新的功能。此外,用于处理和分析的所有参数都存储在“mass_data”对象中,这使得跟踪先前的步骤和参数成为可能。简而言之,“mass_dataset”类提供了一种管理和处理代谢组学数据的简单方法,它为高度可重复、稳健和可扩展的分析框架奠定了基础。
与其他工具的联合使用
为了利用现有工具,我们开发了几个函数,可以将“mass_dataset”转换为其他工具所需的其他数据结构,因此用户可以轻松地将tidyMass与其他工具集成到自己的分析流程中。目前,tidyMass使用XCMS用于取峰和分组,但是我们开发了可以转换mzMine和MS-DIAL生成的数据格式的功能,因此,用户可以从其他工具直接导入成“mass_dataset”,然后使用tidyMass进行后续处理。其他的包括,"SummarizedExperiment", “mzTab-M”格式等,这使得tidyMass拥有与其他工具的高度互通性。
tidyMass的整体工作流程
TidyMass提供了一系列函数,将“mass_dataset”作为输入数据格式来执行LC-MS数据分析的整个流程。整个流程从massConverter 开始,它将来自不同供应商的MS原始数据转换为其他格式。接下来,通过基于XCMS7的massProcesser包对原始数据进行处理、峰值提取,然后通过massCleaner包执行数据清理。在下一步中,metID包用于代谢物注释。所有的统计分析都可以使用massStat包完成。最后,使用metPath包进行通路富集分析。在工作流程的任何步骤中,massQC软件包都可用于评估数据质量。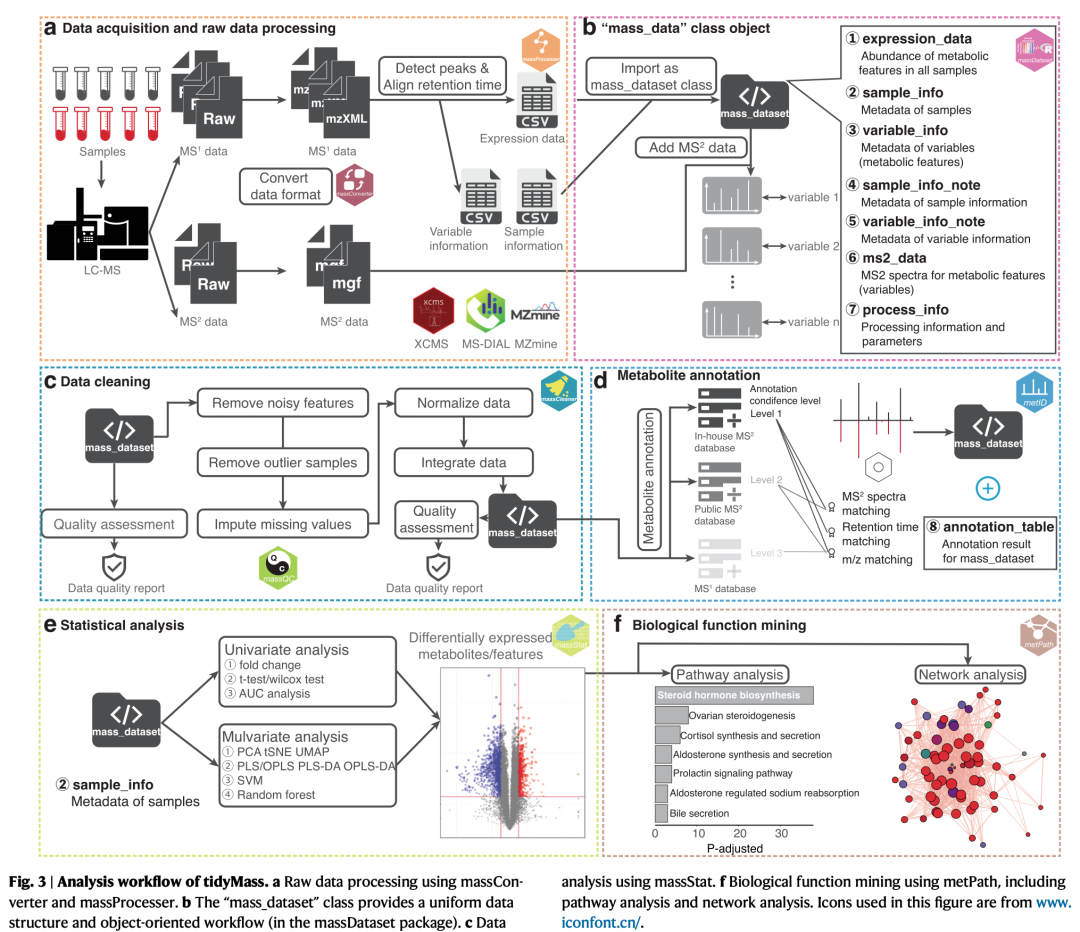
使用tidyMass进行可重复性分析
数据共享和可重复分析对于避免有偏倚的结果至关重要。TidyMass旨在从两个方面实现可重复性和透明度。首先,面向对象的类使得共享数据和跟踪处理信息变得容易。其次,通过统一的数据结构和模块化设计,用户可以将所有处理和分析步骤以一种集成的方式无缝结合在一个代码脚本中(例如,Rmarkdown、notebook)。此外,所有步骤都是可选的,执行顺序是可定制的,这意味着用户可以根据自己的实验设计和目标创建和优化定制的可共享和可重复的管道。此外,随着 docker 技术在可重现分析中越来越流行,我们还提供了 tidyMass的docker版本,包含R/Rstudio环境和所有tidyMass包,这使得用户可以共享所有代码,数据,甚至是基于 tidyMass 的分析环境。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









