







题目:Dissecting the role of the human microbiome in COVID-19 via metagenome-assembled genomes
发表杂志:Nature Communication
发表时间:2022年9月6日
第一作者:Shanlin Ke
通讯作者:YangYu Liu
第一单位:Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School
影响因子:17.69
DOI:10.1038/s41467-022-32991-w
原文链接:https://www.nature.com/articles/s41467-022-32991-w
- 摘要 -
COVID-19是近年来大规模流行的呼吸道疾病,并通常伴有胃肠道症状。然而,我们对于人类微生物组(尤其是肠道微生物)和COVID-19之间的关系知之甚少,这很大程度上是由于以前的大多数研究未能提供高分类学分辨率来鉴定可能与SARS-COV-2感染相互作用的微生物。在这里,我们使用了二代测序配合以组装和分箱策略,重建来自六个独立队列中514个鼻咽和粪便样品中的宏基因组组装基因组(MAGs)。我们总共重建了11584个菌株水平的中等和高质量MAG,并获得了5403个非冗余MAGs(nrMAGs)。我们发现,COVID-19患者的肠道微生物组中许多物种的丰富度显著降低。肠道微生物组的特征可以准确地将COVID-19病例与健康对照区分开,并预测COVID-19的进展。此外,我们确定了一组nrMAGs在COVID-19的临床表现中的功能途径,这些功能途径可能与SARS-COV-2感染相互作用。最后,证明了我们的研究的主要发现可以在三个独立的群组中得到很大的验证。结果强调了将人类肠道微生物组纳入我们对SARS-COV-2感染和疾病进展的重要性。
- 研究背景 -
COVID-19能够引起严重的急性呼吸道综合征2(SARS-COV-2)引起的呼吸道疾病。COVID-19的临床表现范围广泛,包括无症状或轻度疾病,伴有咳嗽和发烧,严重肺炎并伴随有多器官衰竭和急性呼吸窘迫综合征(ARDS),这也是其高致病率和高死亡率的主要。现有研究发现,很大一部分COVID-19患者至少有一种胃肠道(GI)症状,例如腹泻、呕吐或腹痛。据报道,在中国73例SARS-CoV-2感染的住院患者中,53.4%的患者在感染后第1天至第12天的粪便样本中经检测呈SARS-CoV-2阳性。并且更重要的是,在超过20%的感染患者中,即使在呼吸道和/或痰液样本没有检测到病毒之后,他们的粪便样本仍然呈病毒阳性。甚至在某些情况下,粪便中的病毒载量甚至高于咽拭子中的病毒载量。所有这些结果表明,胃肠道可能是SARS-CoV-2感染的除肺部以外的另一重要部位。迄今为止,基于16S rRNA基因测序的几项研究表明,COVID-19患者的人类上呼吸道和肠道微生物组发生了显著的的变化。但16S rRNA没有提供捕获足够的序列变异以区分密切相关的分类群所需的分类分辨率。基于全/宏基因组(WMS)测序的研究通过将短宏基因组读数映射到参考基因组数据库,探索了人类微生物组与SARS-CoV-2感染之间的联系。尽管对WMS测序数据的分析提供了比16S rRNA基因测序数据分析更多的信息,但基于参考基因组数据库的现有研究受到这些数据库的限制和偏见的影响,无法表征没有密切相关的培养代表的微生物。
通过从头组装和分档来重建宏基因组组装的基因组 (MAG),允许回收尚未分离和培养的微生物的基因组。该策略已被几项研究采用,以提供对人类健康和疾病至关重要的微生物种群的基因组见解。在这项研究中,我们应用了先进的宏基因组组装和分档策略,直接从COVID-19患者和对照组的微生物组样本中重建微生物群体基因组(图1)。我们的主要目标是构建一个与COVID-19相关的宏基因组目录,以识别可能与SARS-COV-2感染的临床表现相关的新型分类群和菌株水平差异。我们的研究结果表明,人类微生物组与SARS-CoV-2感染的关联具有前所未有的高分类分辨率。更重要的是,我们的研究提供了一个独特的资源,可以直接调查COVID-19相关微生物菌株的基因组含量,并为更有针对性的后续研究提供依据。
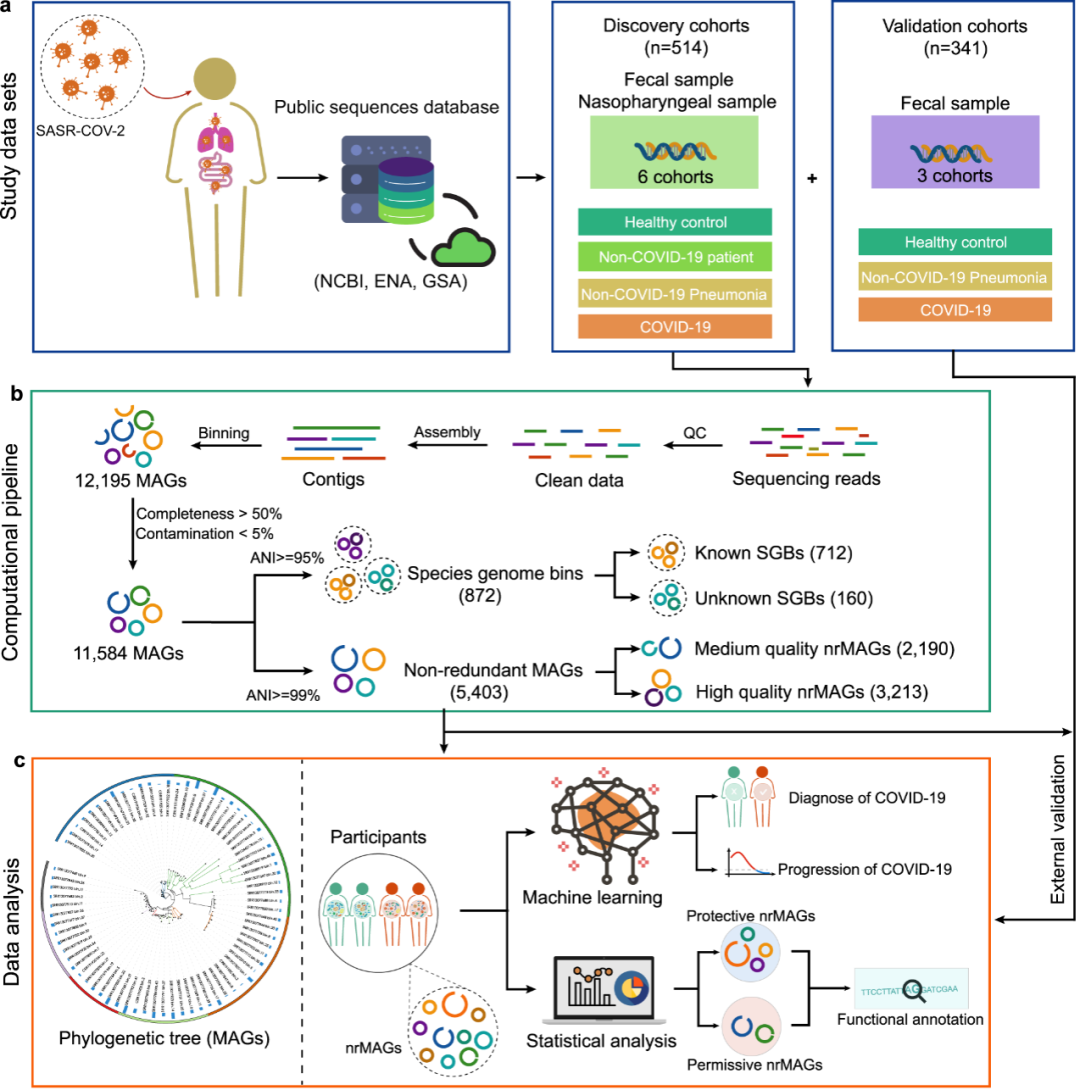
图1:实验设计
a.为了通过宏基因组组装基因组(MAGs)了解人类微生物组与COVID-19之间的关系,我们分别收集了关于发现和验证队列的514(6个队列)和341个(3个队列)宏基因组测序数据。这些微生物组样本包括来自COVID-19和非COVID-19对照组的粪便和鼻咽样本。b.根据发现队列的宏基因组测序数据共构建了11,584个MAG(≥50%完整性和≤5%污染率)。重建的MAG首先聚集在872个种水平的基因组箱(species-level genome binning)(95%的ANI水平)。在基因组分类数据库(GTDB)中含有至少一个参考基因组(或宏基因组组装的基因组)的SGB被认为是已知的SGB。然后,根据99%的ANI,将重建的MAG去冗余为5403个非冗余MAG(nrMAGs,菌株水平)。将5403 nrMAG分为中等质量的MAG(50%≤完整性<90%和≤5%污染率)和高质量MAG(完整性≥90%和≤5%污染)。c. nrMAGs的系统发育树是使用PhyloPhlAn构建的。我们将随机森林机器学习模型与nrMAG结合使用来诊断COVID-19并预测COVID-19的进展(RT-qPCR阴性结果的日期)。COVID-19的许可性和保护性nrMAG由GMPT方法确定。nrMAGs的基因组使用Prokka和微生物注释器进行功能注释。
- 主要结果 -
① 构建的COVID-19高质量微生物基因组目录
在质量控制之后,我们对来自发现队列的微生物组样品进行了宏基因组组装和分箱,总共获得了12195 MAGs,为了标准化所有数据集的基因组质量,我们使用了≥50%基因组完整性和≤5%污染的阈值。产生 11,584 个 MAG [平均完整性 = 87.55%,平均污染 = 0.99%,平均基因组大小 = 2.6 兆碱基 (Mb),平均 N50 = 61.8 千碱基 (kb)。为了获得物种级微生物群落的视图,我们首先将11584个MAG组织到物种水平的基因组箱(SGBs)中,ANI(平均核苷酸同一性)阈值为95%,总共产生了872个SGB,其中160个(18.35%)SG代表了没有任何基因组分类数据库(GTDB)可用基因组的物种,并被定义为未知的SGB。
为了评估最高质量的代表性基因组,我们在ANI阈值为99%的情况下对11584个MAG进行了重复,从而产生了最终一组5403个非冗余MAG(nrMAGs),具有菌株水平的分辨率。比较MAGs的分布发现,每个非COVID-19微生物组样本的总MAG和nrMAG率相对高于COVID-19微生物组样本,因为21.40%的非COVID-19微生物组样本占总MAG的31.32%和38.94%的nrMAG,如图2所示。
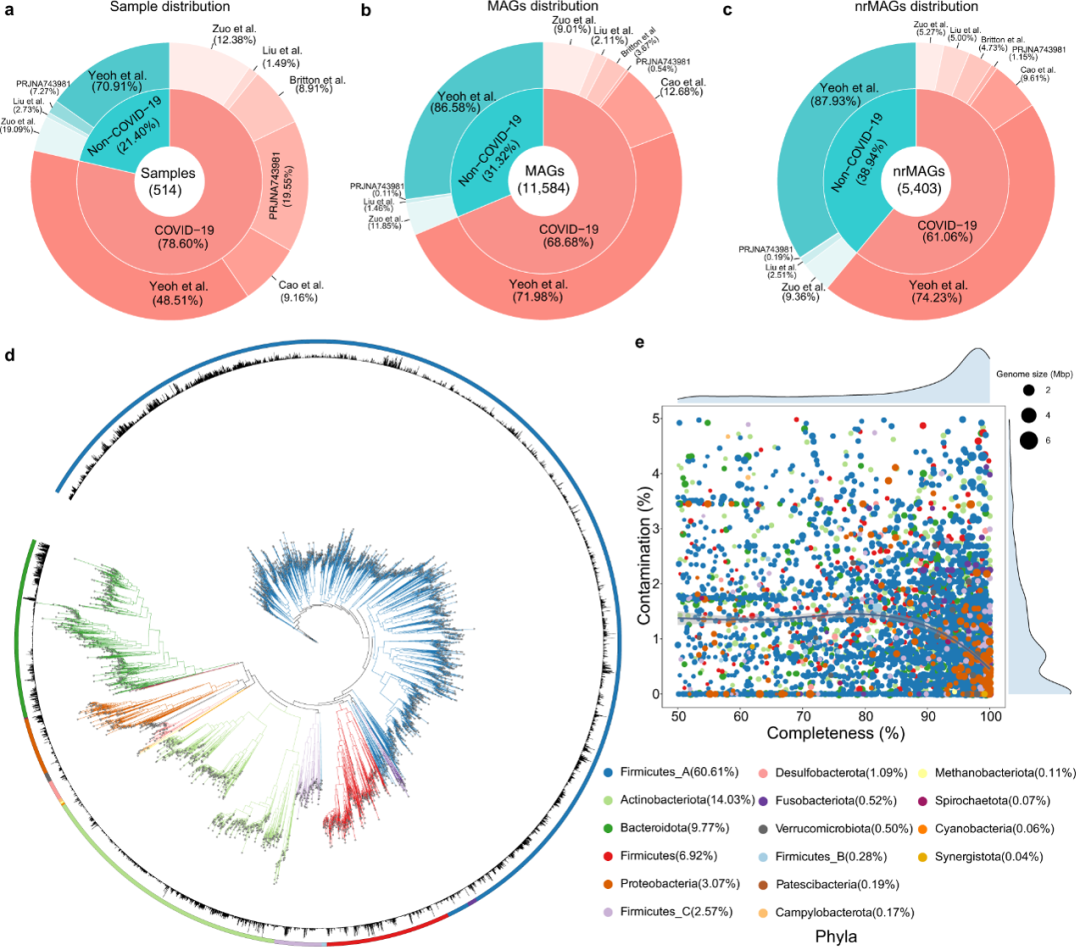
图2:从发现队列中514个与COVID-19相关的宏基因组学测序数据重建MAG
a. 不同数据集和疾病状况之间的样本分布。b 从不同数据集和疾病状况中恢复的MAG数量。c. 从不同数据集和疾病状况中恢复的nrMAGs数量。d. 使用系统发育系统发育构建的 nrMAG。外循环和进化枝的颜色代表门,周期内的条形图代表所有微生物组样品的平均相对丰度。e. 完整性和污染在nrMAG上的分布以及点的颜色代表门。点的大小代表nrMAG的基因组大小。
② COVID-19患者人体微生物组的改变
以前的研究表明,SARS-COV-2感染与人类肠道的α多样性有关。我们首先在我们的发现队列中调查了SARS-CoV-2感染是否与nrMAG水平的人类微生物组的α多样性相关。比较了COVID-19患者和非COVID-19对照组的α多样性测量(即丰富度和香农指数),如图3所示,有趣的是,我们发现COVID-19患者肠道微生物组的α多样性总体上低于验证队列中非COVID-19对照组的α多样性,COVID-19和非COVID-19对照组患者之间的肠道微生物群落结构存在显着差异(PCoA)。同时,疾病严重程度较轻的COVID-19患者在其肠道微生物组中表现出显着更高的Shannon多样性(图3e)。并且,COVID-19患者康复后(通过RT-qPCR对SARS-CoV-2呈阴性)的nrMAGs组成与非COVID-19对照组相比与COVID-19患者在恢复前显着不同。
为了了解疾病严重程度与COVID-19患者肠道微生物组的短期变化之间的关系,我们追踪了与疾病严重程度相关的每个个体中微生物组的变化。有趣的是,疾病严重程度较轻的COVID-19患者在肠道微生物组中表现出较低的时间变异(通过纵向微生物组样本的Bray-Curtis差异性量化,图3h)。在较轻的疾病严重程度组中,肠道微生物组样品的时间变异较低,部分原因是这些组中纵向微生物组样品中常见nrMAG的比例较高(图3i)。
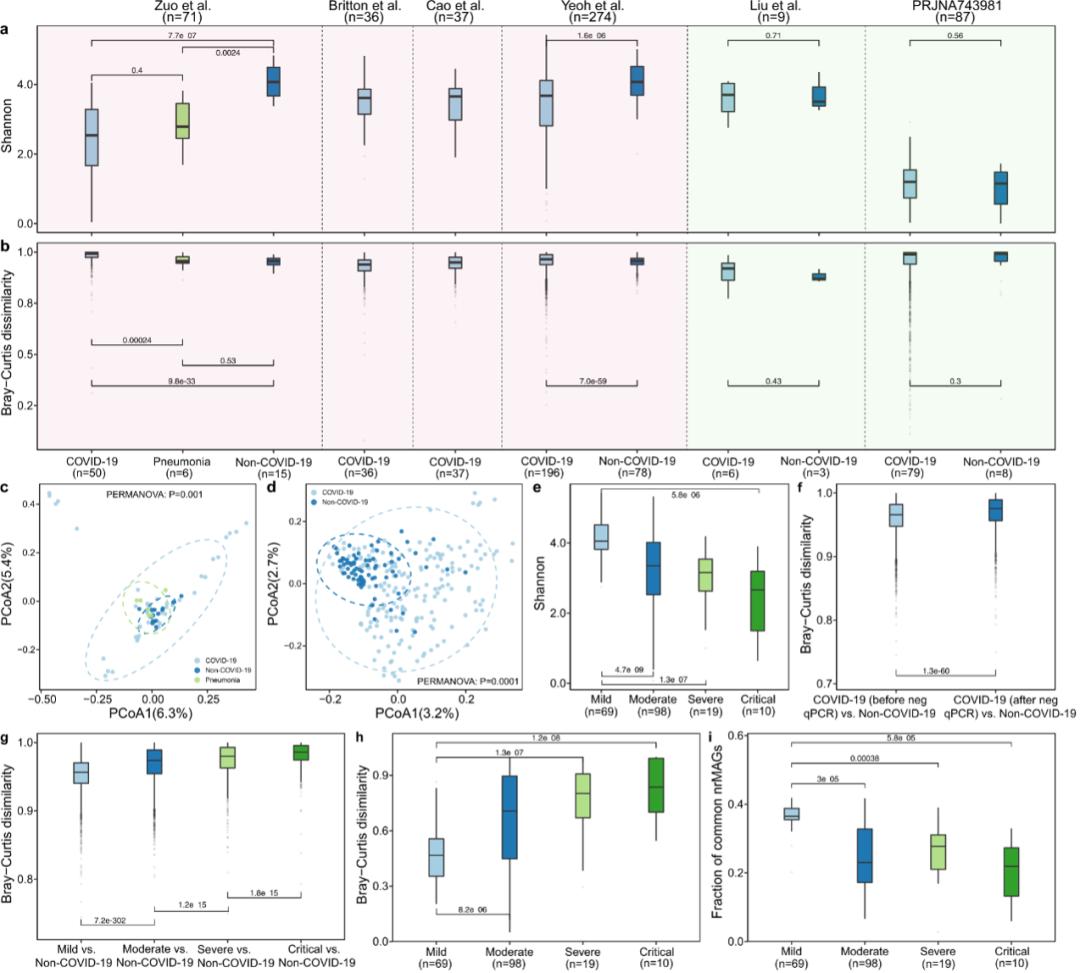
图3:发现队列中人类微生物组的COVID-19相关变化
香农多样性(a)和组中布雷 - 柯蒂斯不同性(b)在nrMAG水平上的人类微生物组来自每项研究。每个面板的背景色(a,b)代表来自人体肠道(浅红色)或鼻咽(浅绿色)的微生物组样品的来源。主坐标分析(PCoA)图基于Zuo等人(c)和Yeoh等人(d)研究的微生物组成的Bray-Curtis差异性。所有PERMANOVA测试都是用基于9999排列进行的。e. 来自Yeo等人研究的不同疾病严重程度组的香农多样性。f. 肠道微生物组的箱形图 Bray-Curtis 健康对照组与COVID-19患者在鼻咽抽吸物或拭子之前或之后的差异通过RT-qPCR检测SARS-CoV-2阴性。g. 健康对照组和来自不同疾病严重程度的组的COVID-19患者之间的肠道微生物组的箱形图。h. 来自Yeo等人研究的不同疾病组的单个微生物组的时间变化的盒子。i. Yeoh等人的研究随着时间的推移,COVID-19患者的常见nrMAGs的比例。
③ COVID-19患者失去了多种微生物的菌株
为了探索同一物种内的菌株水平多样性是否与COVID-19有关,我们首先根据GTDB分类信息将所有nrMAGs分组到物种水平。对于每个物种,我们计算了所有微生物组样品的菌株丰富度(即其nrMAGs的数量)。具有最高菌株丰富度的前30种微生物物种在发现和验证队列之间高度重叠(图4a和b)。值得注意的是,我们发现,与非COVID-19对照组相比,COVID-19患者在发现(图4c,d)和验证队列方面都失去了许多种水平上的菌株,包括Bariatricus comes、Blautia_A obeum、Blautia_A wexlerae、Dorea formicigenerans、Faecalibacterium prausnitzii_D、Faecalibacterium sp900539945、和Fusicatenibacter saccharivorans。这些结果与nrMAG级别的α多样性分析高度一致,即COVID-19患者的发现队列中非COVID-19对照组(即Zuo等人和Yeoh等人)发现的nrMAGs数量明显低于非COVID-19对照组。
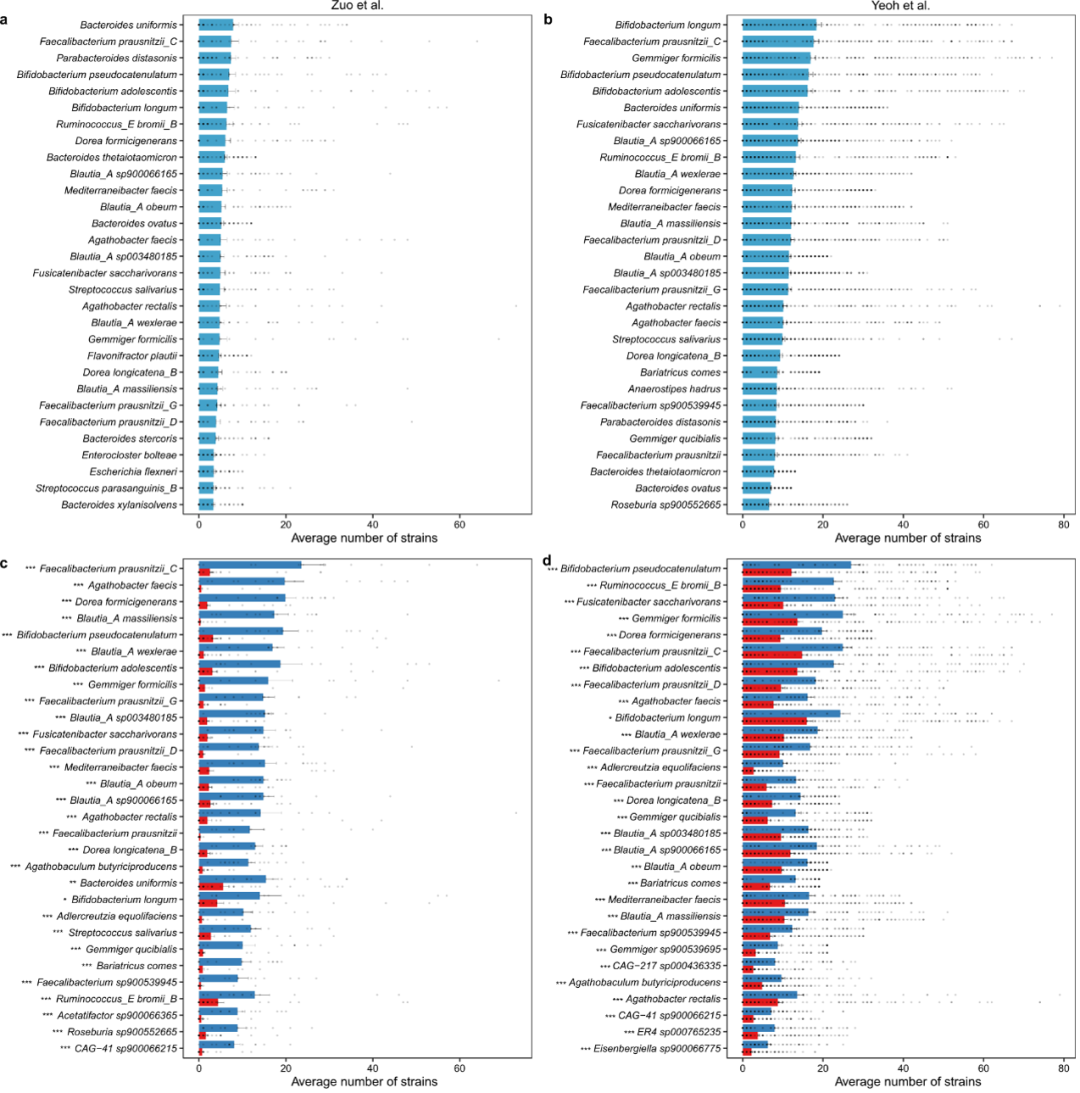
图4:两个发现队列中微生物物种菌株丰富度的COVID-19相关变化
a和b. 从Zuo等人(样本数量n = 65)和Yeoh等人(b样本数量n = 274)的研究中确定的具有最高菌株丰富度(即nrMAGs数量)的前30个物种。c和 d. 从左等人(c)和Yeo等人的研究中确定的非COVID-19(蓝色)和COVID-19(红色)样本之间菌株丰富度变化最高的前30个物种。数据以均值±均值标准误表示。P值通过双侧威尔科克森-曼-惠特尼检验计算(ns不显著,*P <0.05; **P < 0.01,***P < 0.001)
④ nrMAG 可准确分类 COVID-19 患者和非 COVID-19 对照组
以前的研究已经证明,使用属或种级分类学特征对SARS-CoV-2感染进行基于微生物组的分类具有诊断潜力。为了测试nrMAG级别的肠道微生物组成是否可以区分COVID-19患者和非COVID-19对照组,我们在两个数据集上构建了随机森林分类模型。我们应用了两个指标来量化分类性能:AUROC(工作特性曲线下的面积)和AUPRC(精度-召回曲线下的面积)。我们发现nrMAG可以准确地检测出COVID-19,平均AUROC和AUPRC值分别为0.981和0.971。与COVID-19相关的主要特征包括:Adlercreutzia equolifaciens, Blautia_A sp003471165, Eisenbergiella sp900066775, Eubacterium I, Gemmiger sp900539695 和 Romboutsia timonensis。
⑤ nrMAG能准确预测新冠肺炎疫情的进展
接下来,我们调查了nrMAGs与COVID-19进展之间的关系。为了探索这种关联,我们采用随机森林回归模型,利用Yeoh等人的数据预测RT-qPCR结果为阴性的日期。这种方法表明,nrMAGs很好地预测了RT-qPCR结果为阴性的日期(Pearson相关性0.425,P值= 1e − 45,图5a)。在前30个(基于均方误差增加百分比)中最重要的nrMAGs包括Citrobacter freundii, Enterocloster sp900543885, Citrobacter portucalensis, Parabacteroides distasonis 和 Veillonella parvula。我们还发现了一些来自众所周知的机会性病原体的nrMAGs:MAG02074 (Klebsiella quasivariicola), MAG03769 (Klebsiella pneumoniae), 和 MAG02080 (Escherichia coli_D)。
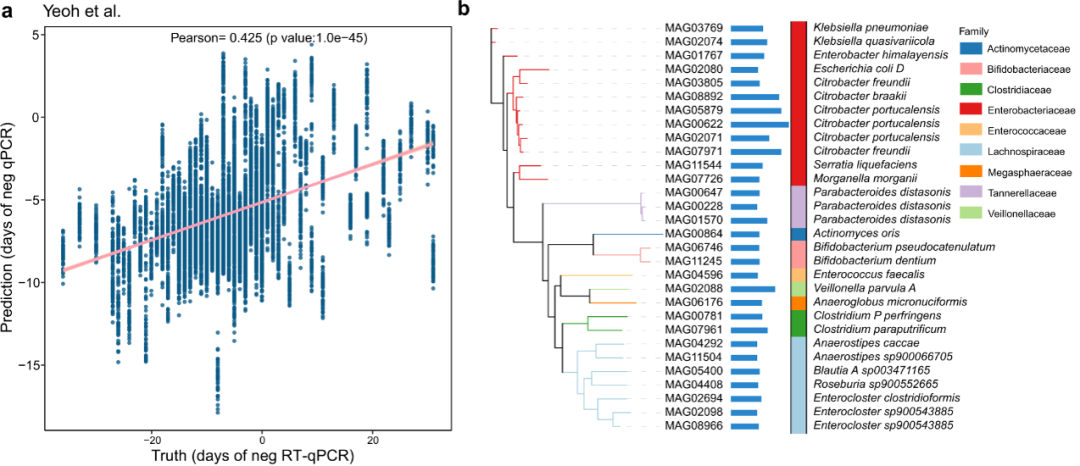
图 5:基于 nrMAG 的机器学习模型预测了 COVID-19 的进展
a. 随机森林回归模型上负RT-qPCR结果的真实日期和预测日期之间的皮尔逊相关系数。b. 与预测业绩有关的前30名重要NrMAG。回归中每个特征的重要性通过均方误差增加百分比(%IncMSE)来量化。水平条形的长度表示平均%数。竖线的颜色表示nrMAG的分类信息。
⑥ 确定针对COVID-19严重程度的潜在的许可和保护性nrMAG
为了进一步表征人类肠道微生物组与COVID-19之间的关系,我们应用了广义微生物表型三角测量(GMPT)方法(图6a),超越了标准的关联分析。使用这种方法,所有成对差异丰度分析都产生了至少两个成对比较中存在的总共644个差异丰度nrMAG。为了了解这些候选nrMAGs与COVID-19之间的潜在关系,我们随后计算了不同表型组中nrMAGs的平均相对丰度与COVID-19严重程度评分(例如,非COVID-19健康对照:0;轻度:1;中度:2,重度:3和临界:4)之间的斯皮尔曼相关系数。那些具有正(或负)斯皮尔曼相关系数的差异丰度nrMAG是COVID-19的潜在允许(保护性)nrMAG。基于所有成对比较的频率(≥6)(n = 10),我们总结了图6b中的GMPT和补充数据2的结果。该分析共确定了74个与SARS-CoV-2感染相关的nrMAG,包括8个允许性(Spearman相关性 > 0)nrMAGs,63个保护性(Spearman相关性<0)nrMAGs和3个中性nrMAG(Spearman相关性= 0)。
从这些允许性nrMAGs(图6b)中鉴定出多个具有高度相似基因组的物种,包括Enterocloster bolteae (3 nrMAGs), Anaeroglobus micronuciformis, Hungatella effluvii (3 nrMAGs) 和Enterococcus_B faecium。与之前的研究结果一致,与健康对照组相比,COVID-19患者的肠道微生物组显示出显着更高的Enterococcus faecium。此外,我们发现两种允许物种(即Enterocloster bolteae 和 Hungatella effluvii)的菌株丰富度(nrMAGs数量)与疾病严重程度呈正相关。值得注意的是,与非COVID-19对照组相比,多种保护性物种(例如,Bariatricus comes, Blautia_A obeum, Blautia_A wexlerae, Dorea formicigenerans, Faecalibacterium prausnitzii_D, Faecalibacterium sp900539945和 Fusicatenibacter saccharivorans)在COVID-19患者中失去了许多菌株(图4c,d).大多数保护性微生物物种的菌株丰富度(17/21)与疾病严重程度呈负相关。有趣的是,在Zuo等人的研究中,这些保护性nrMAGs在COVID-19和非COVID-19对照组患者之间也显示出相似的丰度分布。18(图 S18)。这一发现提供了菌株水平的证据,证明肠道微生物分类群可能与SARS-COV-2感染相互作用,并在COVID-19的疾病发作和进展中起潜在作用。
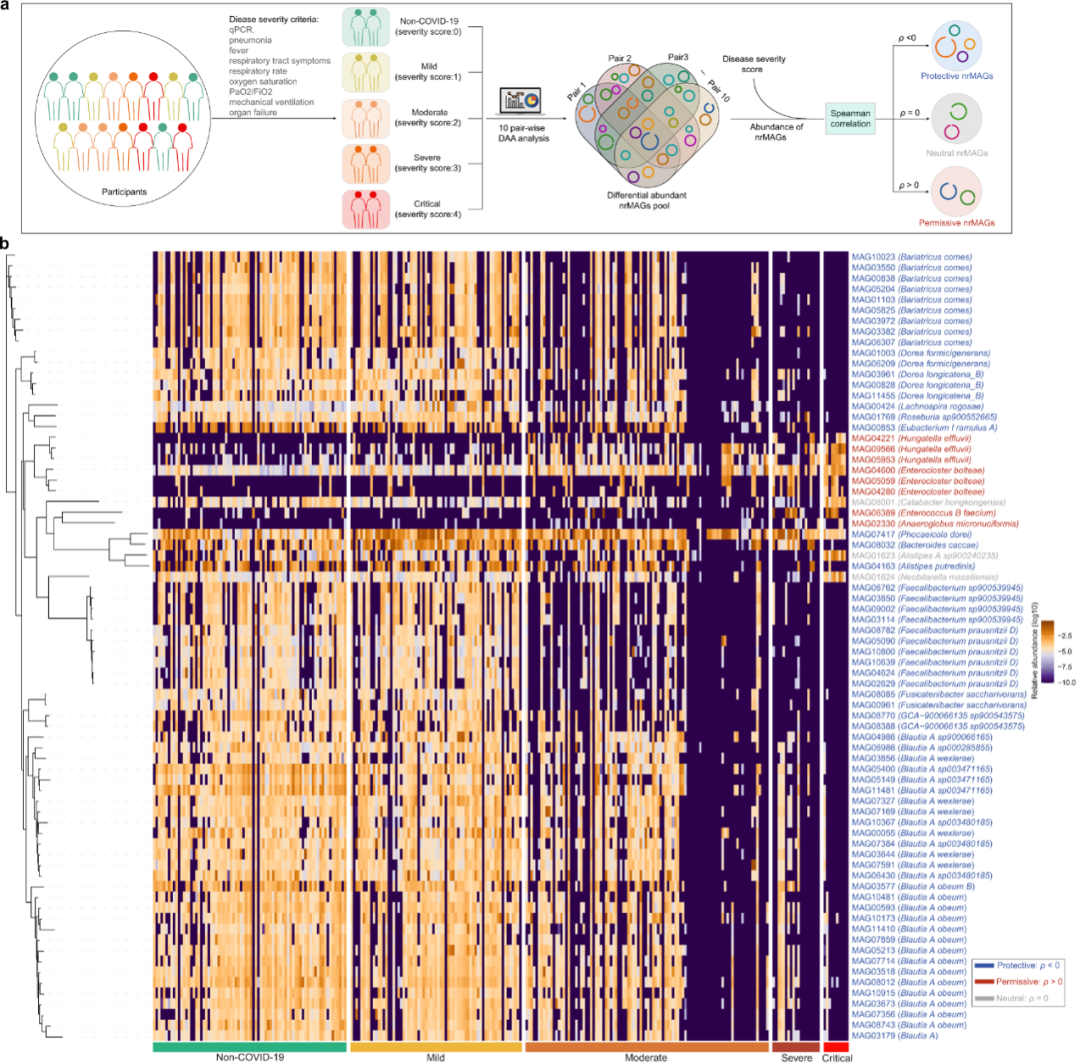
图6:GMPT方法确定的COVID-19的允许和保护性nrMAG
a. GMPT方法的工作流程。Yeoh等人研究的微生物组样本根据疾病严重程度分为五组(即健康对照组,轻度,中度,重度和临界)。对每个可能的成对比较(5组之间的10个成对比较)进行了差异丰度分析。差异丰度nrMAGs根据其在所有成对比较和可微性(降序)中出现的频率进行排名。我们进一步使用nrMAGs的平均相对丰度和疾病严重程度来计算斯皮尔曼相关系数。正(负)斯皮尔曼相关系数 (ρ) 表示 COVID-19 严重程度的允许性(保护性)nrMAG。而长矛兵与0的相关性意味着nrMAGs可能对SARS-CoV-2感染的严重程度是中性的。b. 热图显示了使用GMPT确定的不同疾病严重程度组中允许性、中性和保护性nrMAGs的丰度分布。这些 nrMAG 使用基于基因组分类数据库的 GTDB-Tk 进行分类注释。
⑦ 基因组注释揭示了COVID-19的许可性和保护性nrMAG之间的功能差异
我们接下来研究了允许性和保护性nrMAGs的功能是否有所不同。为了实现这一目标,我们首先使用Prokka注释了允许和保护性nrMAGs的基因组。然后,我们使用MicrobeAnnotator注释并计算 KEGG 模块的完整性。主成分分析显示,允许性nrMAGs和保护性nrMAGs之间的代谢潜力完全不同(图7a,PERMANOVA:P值= 0.0001)。值得注意的是,我们确定了一组KEGG模块,这些模块在允许和保护性nrMAG之间的模块完整性方面存在显着差异(图7b)。例如,与保护性 nrMAG 相比,允许性 nrMAGs 在磷酸戊糖通路(例如 M0004 和 M0006)上显示出更高的完整性。
为了进一步验证磷酸戊糖途径与COVID-19之间的关联,我们对来自病例对照实验设置的两个发现队列中的宏基因组学测序样本进行了功能分析。我们发现COVID-19患者中磷酸戊糖途径(PENTOSE-P-PWY)的丰度显着高于两个发现队列的非COVID-19对照组。这一结果与允许的nrMAGs在戊糖磷酸化途径上显示出显着更高的完整性水平的结果一致。
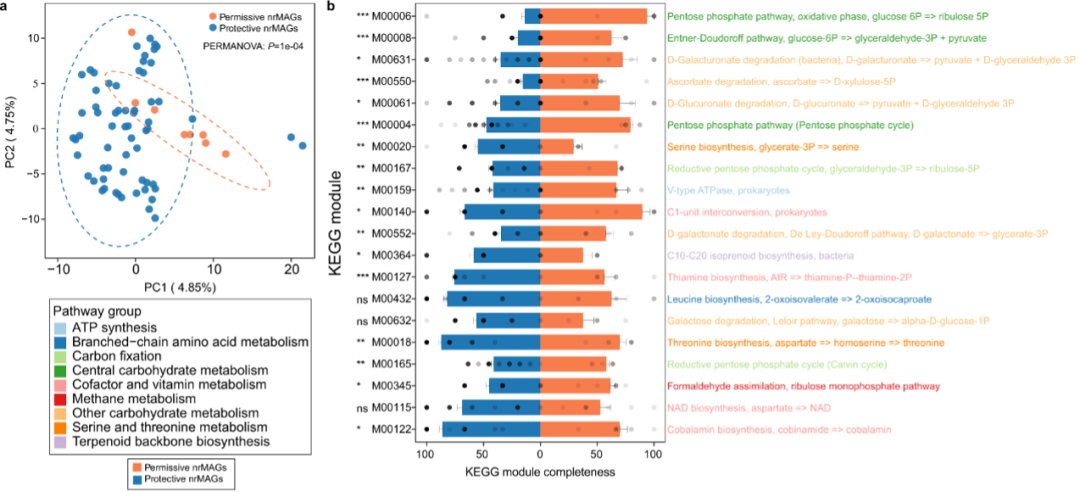
图7:COVID-19允许性和保护性nrMAG的基因组注释
a. 来自允许性(n = 8)和保护性(n = 63)nrMAG的所有基因组的KEGG模块完整性的主成分分析(PCA)。PERMANOVA采用了9999次的排列变化。b. 允许性(n = 8)和保护性(n = 63)之间的前20个差分KEGG模块根据均数差进行排名。数据以均值±均值标准误表示。P值由双侧Wilcoxon–Mann–Whitney检验计算(ns不显著,*P<0.05; **P < 0.01,***P < 0.001)。
- 讨论 -
我们利用了来自多个SARS-COV-2数据集的数百个公开的WMS测序样本,并首次生成了人类微生物组的高质量COVID-19相关基因组基因组目录。我们发现了一个大型基因组目录,代表了人类微生物组的11584个MAG和5403个nrMAG。通过构建这个微生物基因组目录,我们能够提供菌株水平的视角来理解人类微生物组和COVID-19。
通过使用具有不同技术设置的 WMS 测序数据,我们更全面地了解了与 COVID-19 相关的微生物群落。由于不同数据集之间存在固有的差异(例如,年龄,饮食和遗传背景),我们的目标不是跨数据集进行有针对性的比较。重要的是,我们的研究主要集中在两个发现队列和三个验证队列。
以往的研究中将人类微生物组多样性与COVID-19建立联系。在我们的研究中,COVID-19患者的人类肠道微生物组在nrMAGs水平上表现出与非COVID-19健康对照组相比总体上降低了α多样性。具体而言,肠道微生物组的丰富性(nrMAG数量)在发现和验证队列中显示出COVID-19和非COVID-19样本之间具有一致的差异。值得注意的是,在发现和验证队列中,与非COVID-19对照组相比,我们首次发现COVID-19患者在某些微生物物种中丢失了许多菌株(nrMAG)。这些发现表明,SARS-CoV-2感染与特定物种可能引起的总菌株的减少有关。此外,一些失去的物种也被GMPT鉴定为保护性微生物物种。有趣的是,我们的分析发现,与康复前的COVID-19患者相比,康复后的COVID-19患者(通过RT-qPCR对SARS-CoV-2呈阴性)与非COVID-19对照组的差异更大。这一发现支持了COVID-19患者的肠道微生物组在从COVID-19中康复后可能无法立即恢复到相对健康的状态的可能性。鉴于许多从SARS-CoV-2感染中恢复过来的患者经历了长时间的COVID-19症状,我们假设长期疾病症状可能与肠道微生物组的变化有关,但这需要进一步探索。
使用机器学习模型,我们证明了nrMAG级别的肠道微生物组特征可以从健康对照中准确检测到COVID-19。特征中的关键微生物菌株可能在COVID-19的发病机制中发挥重要作用。我们还证明,从特定发现队列中鉴定出的肠道微生物组特征可以跨分离队列诊断COVID-19,而与宿主遗传学和环境因素对肠道微生物组的影响无关。一些关键的微生物物种可能在SARS-CoV-2感染的病理生理学中发挥非常重要的作用。值得注意的是,这项研究揭示了nrMAGs预测COVID-19患者RT-qPCR结果阴性日期的能力。该分析将重要的nrMAGs中的几种微生物物种与COVID-19的进展联系起来。
之前的一项研究表明,COVID-19阳性患者血清中糖酵解和戊糖磷酸酯途径的某些中间体水平显着升高。此外,早期的一项研究(阿拉伯联合酋长国86名COVID-19患者和57名健康对照组)表明,使用16S rRNA基因测序以及功能预测(PICRUSt)指出,在COVID-19患者微生物组样本上显着上调磷酸戊糖途径。此外,还发现SARS-CoV-2感染与体内戊糖磷酸酯途径调节的变化有关。总之,这些结果表明,特定的微生物(允许性nrMAGs)可能在介导SRAS-CoV-2通过戊糖磷酸途径和芳香族氨基酸进入宿主细胞中发挥作用。然而,有必要进行进一步的机制研究。
目前的研究也有一些局限性。首先,尽管我们纳入了来自COVID-19相关人类微生物组研究的大量猎枪宏基因组测序样本(分别于2021年8月和2022年4月公开了发现和验证队列),但大多数微生物组样本来自中国。通过从不同人群和身体部位收集更多的人类微生物组样本来构建更全面的基因组目录,以揭示COVID-19中人类微生物组的完整景观,可以解决这一限制。其次,尽管我们在统计模型中调整了潜在的混杂因素,但我们无法评估一些协变量,例如:药物,饮食和心理压力,这些都不是公开的。第三,鉴于分箱非细菌基因组具有相当挑战性,我们只从细菌和古菌中回收了MAG,未来其他领域(包括真菌和病毒)的研究目标将在宿主特异性微生物群和COVID-19的背景下提供更全面的视角。尽管我们在这项研究中重建的大多数MAG都具有高质量,但未来旨在恢复微生物完整基因组的研究将进一步增强我们对人类微生物组与SARS-CoV-2感染之间相互作用的理解。最后,需要进行额外的实验来评估候选许可和保护性nrMAG在COVID-19进展中的作用。然而,鉴于人类肠道微生物组中仍然存在大量未培养的多样性以及注释基因和参考基因组的缺乏,拥有高质量的基因组目录可大大提高基于宏基因组的COVID-19研究的分辨率和准确性。因此,所提出的基因组目录代表了机械地理解人类肠道微生物组在SARS-CoV-2感染中的作用的关键一步。
总之,我们在这里介绍在COVID-19和非COVID-19对照组中使用宏基因组的组装和无参考分箱的基因组的首次构建。我们的研究结果支持SARS-CoV-2感染与人类肠道微生物组之间的密切联系,并且我们证明这项研究的主要发现可以在独立队列中得到很大程度的验证。这些对人类微生物组和COVID-19以及基因组背景中宏基因组菌株水平关系方面的见解将构成未来研究的基础。
- 通讯作者 -

哈佛大学医学院
布莱根女子医院
刘洋彧
副教授,副研究员
刘洋彧,哈佛大学医学院副教授,布莱根女子医院副研究员。刘洋彧于2009年在伊利诺伊大学厄巴纳-香槟分校获得物理学博士学位,论文主题是无序磁体的相变研究。之后,他在东北大学复杂网络研究中心先后担任博士后和研究助理教授。他在东北大学研究的主要课题涉及结合控制论、网络科学和统计物理等工具解决与复杂系统控制相关的基本问题。他在复杂网络系统的可控性和可观察性方面的工作被列为Nature的封面故事、PNAS的封面故事,并被包括Nature、Science、Science News、Science Daily、Wired 等在内的广泛媒体报道。2013年他加入哈佛大学医学院和布莱根女子医院。他目前的研究工作侧重于从群落生态学、网络科学,控制论和机器学习的角度研究微生物组。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









