






点击蓝字 关注我们
MIST:基于NGS数据的微生物鉴定和溯源分析平台

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.146
● 2023年11月2日,上海市食品药品检验研究院、上海美吉生物医药科技有限公司和上海市食品药品包装材料测试所在 iMeta 在线联合发表了题为 “MIST: A Microbial identification and source tracking system for Next-generation sequencing data” 的文章。
● 本研究开发了一体化微生物鉴定系统MIST(http://syj.i-sanger.cn),并为 MIST 开发了一个可公开访问的 Web 分析平台,用户可在线开展数据分析工作。
● 第一作者:宋明辉、韩畅、刘林梦
● 通讯作者:任一(upforpunkin@gmail.com)、韩畅(goodluckhc@163.com)、秦峰(sifdcqinf@163.com)
● 合作作者:杨美成、李琼琼、范一灵、高豪、张丹
● 主要单位:上海市食品药品检验研究院、上海美吉生物医药科技有限公司、上海市食品药品包装材料测试所
亮 点
● MIST是基于NGS数据的集成的交互式微生物鉴定和溯源分析系统;
● MIST满足食品药品质量控制中微生物鉴定和污染物调查的需要;
● 系统可直接连接NGS仪器;
● 友好的图形用户界面,助力用户轻松完成数据挖掘。
摘 要
国家药典委员会发布了《微生物全基因组测序技术导则》(草案),旨在规范微生物WGS的方法流程和技术指标,确保测序鉴定的准确性。在上述《导则》的基础上,我们开发了一体化微生物鉴定系统MIST,可以满足食品药品质量控制中微生物鉴定和污染调查的需要。MIST集成了三种分析流程:基于16S/18S/ITS扩增子的微生物鉴定、基于WGS的微生物鉴定和基于SNP的微生物溯源追踪。MIST可以分析多种格式的序列数据,如Fasta、BCL、Fastq等。MIST可以连接到高通量测序仪器,直接获取测序数据。此外,我们为 MIST (http://syj.i-sanger.cn) 开发了一个可公开访问的 Web 分析平台,用户可在线开展数据分析工作。
视频解读
Bilibili:https://www.bilibili.com/video/BV1W94y1H7JY/
Youtube:https://youtu.be/L1MsO1ElWXM
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
微生物鉴定对临床、流行病学、食品和药学研究具有重要价值。通常,基于微生物的形态、物理和生化特性可对微生物进行鉴定。然而,为数众多的原核微生物难以使用传统的纯培养技术获得微生物克隆,因此无法通过上述方法进行检测。这些不可培养的微生物具有新型代谢物的潜在来源,是天然代谢网络的重要组成部分。此外,传统的微生物鉴定方法,难以鉴定未知的微生物。高通量测序技术(HTS)的兴起,使基于核酸序列的基因组学方法成为微生物鉴定的常规和有前途的方法之一。基于HTS的方法可分为两类:1)扩增子测序,扩增子测序扩增微生物中的保守序列(例如,细菌的16S rDNA和真菌的18S rDNA/ITS区域),2)全基因组测序(WGS),在微生物纯培养后对微生物的全基因组进行测序。基于16S rDNA的扩增子测序是研究样本中所有细菌的有效方法,因为该区域已被公认为微生物群落鉴定的常规方法。在Ribosomal Database Project(RDP)和SILVA等大型数据库中,已经积累了大量表征良好的16S rDNA序列。扩增子测序的一个主要局限性是缺乏对近源物种的区分。基于WGS技术的微生物鉴定提供了更高的鉴别能力,并允许在物种甚至菌株水平上鉴定细菌。它还为研究功能基因(如抗生素耐药基因(ARGs)和毒力因子基因提供了强有力的方法。此外,多位点序列分型(MLST)和单核苷酸多态性(SNP)能够追踪从不同生境分离到的遗传密切相关细菌的来源。这种分析使基于WGS的应用能够在多个领域得到应用,例如法医调查、菌株鉴定和疫情追踪。越来越多的微生物鉴定平台和工具被广泛应用,例如BacWGSTdb、ImageGP 、CGE、Qiime2、EasyAmplicon、GCType和rANOMALY。每个平台都有其独特的优势和局限性。例如,BacWGSTdb 提供基于 MLST 和基于全基因组的细菌基因分型,但只接受组装基因组文件作为输入。CGE 为基于基因组的 ARG 和 VFG 表型、系统发育和注释提供了分析工具。但是,由于缺乏集成的后端,用户需要将他们的数据分别上传到这些工具中。此外,所有基于Web的工具都需要快速且一致的互联网连接来上传原始序列文件,其大小可能为数百至数千MB。随着NGS技术的发展,下游生物信息学分析具有挑战性,需要开发更多的数据挖掘软件和系统。
我们开发了一个用于微生物分类和鉴定的系统。它利用扩增子测序数据有效地分析不可培养微生物,基于WGS数据可准确地进行基因分型并完成ARG和VFG注释,此外平台还实现了基于SNP的菌株溯源分析。
结 果
系统概述
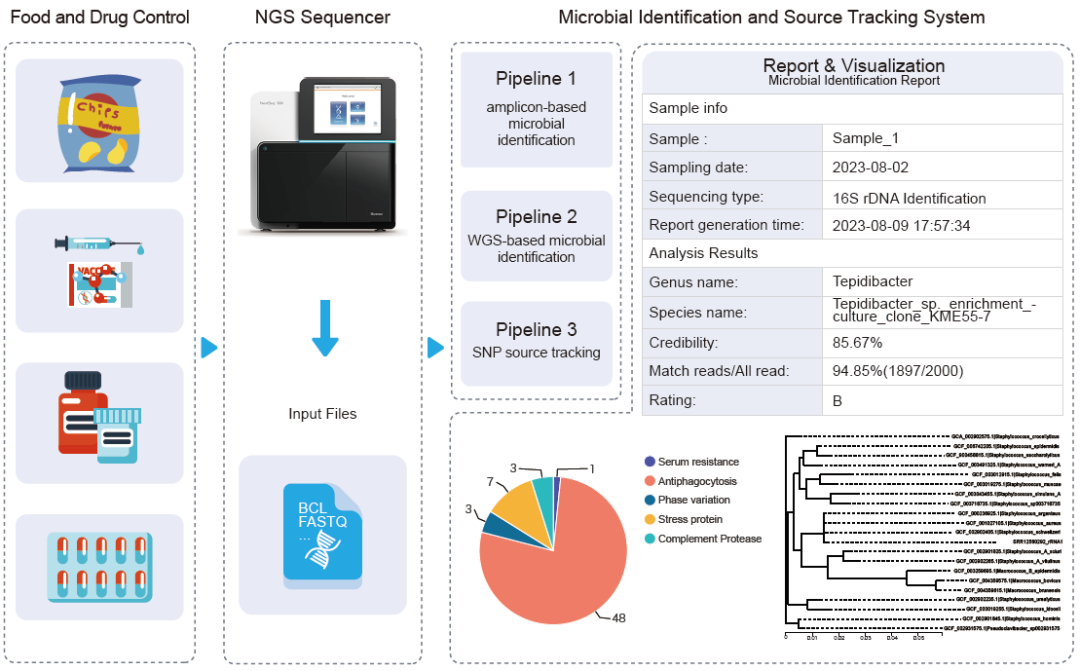
该系统由三个分析流程组成:1.基于扩增子的微生物鉴定,如16S rDNA/18S rDNA/ITS基因,2.基于WGS的微生物鉴定,3.基于SNP的溯源分析。用户需选择Illumina测序仪生成的BCL或fastq格式的测序文件,或fasta格式的序列文件(如组装基因组或16S序列等),创建分析任务,设置分析参数,点击提交,前端界面将文件和参数传输至服务器,并触发分析流程的运行(图1A)。该系统为微生物鉴定和功能注释提供了主流的参考数据库(图1B)。此外,配备了数据管理系统,负责监控处理任务和管理数据库,例如输入和输出文件等(图 1D)。用户可以在在线交互式分析报告界面上查看任务结果,并下载结果以供进一步数据挖掘使用(图1C)。
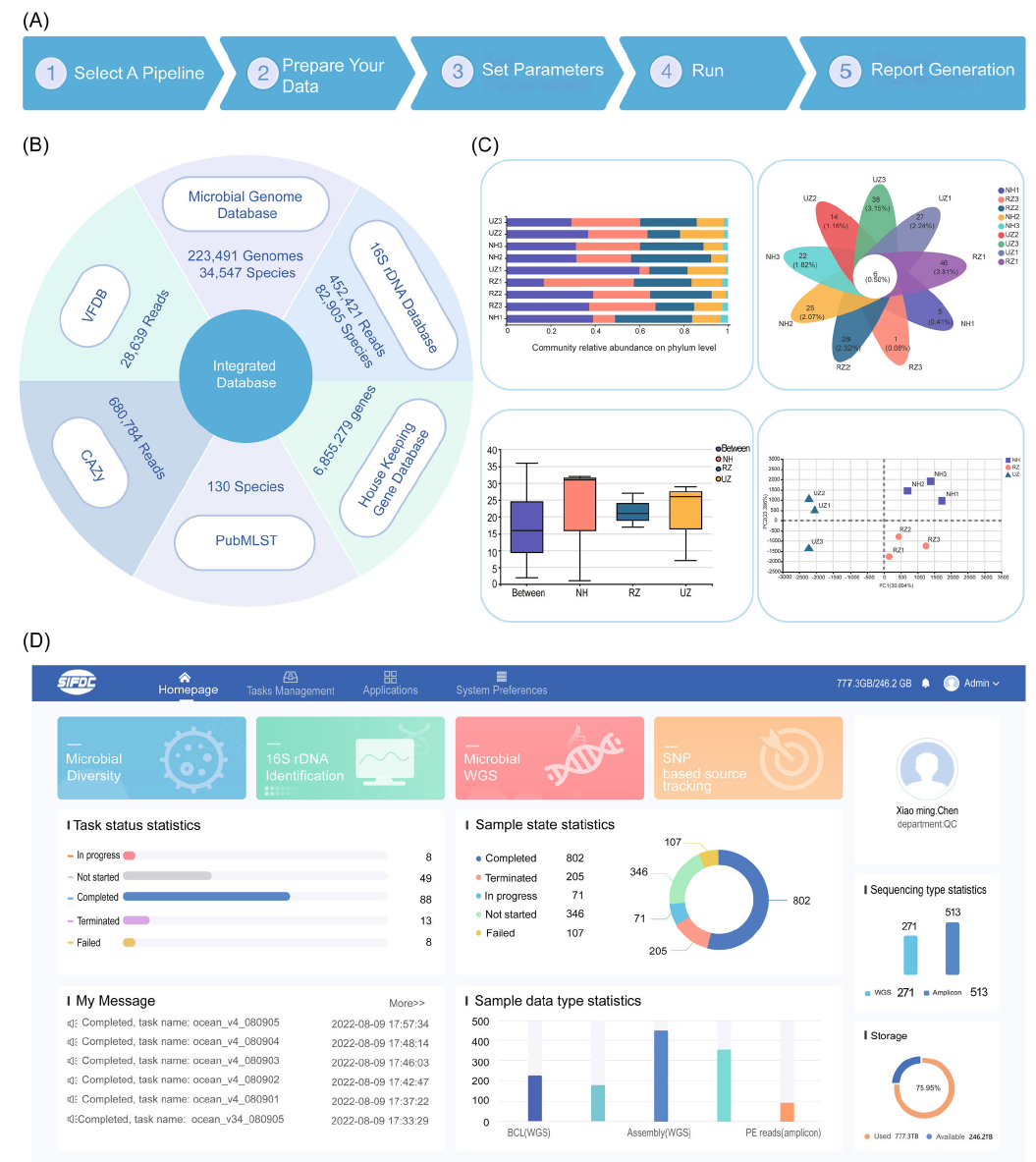
图1. MIST系统概述
(A) MIST的五个数据分析步骤;(B) MIST中整合的物种和功能数据库;(C) 数据可视化举例;(D) MIST 系统主页。
分析流程1:基于16S rDNA/18S rDNA/ITS扩增子的微生物鉴定
该分析流程适用于 16S/18S rDNA 和 ITS 区域的扩增,鉴定可培养或不可培养微生物。该流程包含“质量控制”、“接头序列去除”、“降噪”、“注释和评估”功能参数。简而言之,Fastp v0.23.4 用于执行质量控制,并通过根据质量和长度修剪和过滤读数来清理双端 FASTQ Reads。在50 bp滑动窗口中,截断平均质量得分为 <20的低质量序列,去除短于50 bp的reads,同时去除包含模糊碱基的序列。合并双端reads,然后通过自主编写的Python脚本删除引物,采用vsearch v2.22.1删除重复序列,并通过deblur v1.1.1 去噪。上述过程,生成一组扩增子序列变体 (ASV),每个变体都被视为一个分类单元。然后使用BLASTn v2.11.0将每个ASV与参考基因组数据库进行比对。ASV的分类学注释是通过参考数据库中的最佳命中率匹配来估计的。采用最大似然(ML)方法构建系统发育树。分析流程如图2A所示。
我们从人类肠道、海洋两种不同的类型的环境样中选取了数十种细菌,并基于16S核糖体基因的V3-V4、V4和V4-V5区域生成了相应的测试数据。基于测试数据,测试了扩增子鉴定程序的性能。所有细菌均在属水平上均被正确鉴定。(表 S1)。
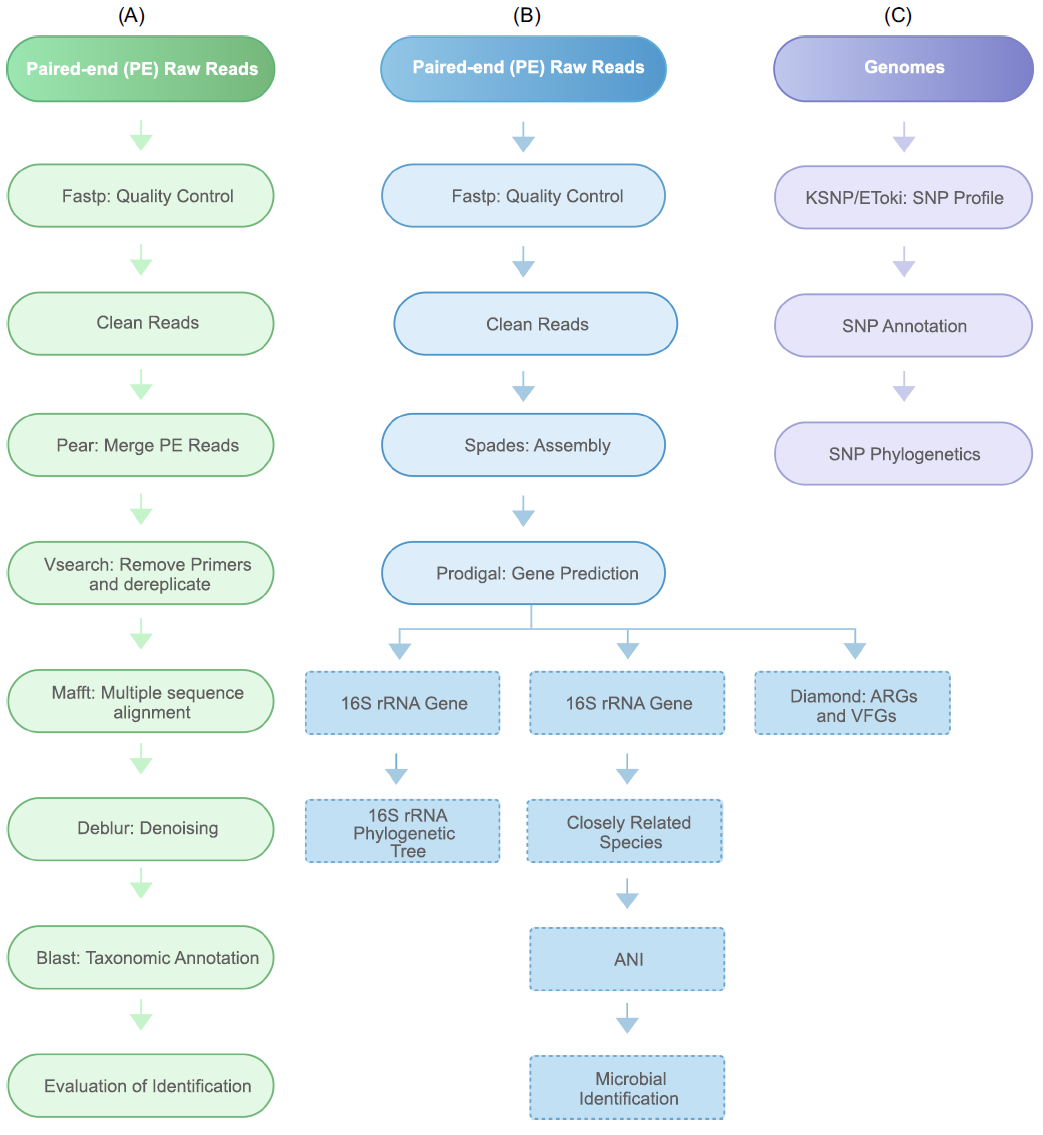
图2. 3个分析工作流的流程图
(A)基于扩增子的微生物鉴定,如16S rDNA/18S rDNA/ITS基因;(B)基于SNP的来源追踪;(C)基于全基因组测序的微生物鉴定。ANI,平均核苷酸一致性; ARGs,抗生素抗性基因; rRNA,核糖体RNA; SNP,单核苷酸多态性; VFGs,毒力因子基因; WGS,全基因组测序。
分析流程2:基于WGS的微生物鉴定
近年来,基于WGS的微生物鉴定已广泛地应用于基础研究和临床诊断。在我们的系统中,我们使用持家基因和平均核苷酸一致性(ANI)来识别微生物物种并推断其与其他物种的系统发育关系。该流程包含六个模块:质量控制、组装、基因预测、ANI计算、注释和MLST。
Fastp 用于质量控制。在组装过程中,使用SPAdes v3.11 组装基因组。对于一些其他微生物污染的样本,使用metaSPAdes v3.10 进行污染的样本组装。BUSCO v5.1 用于评估基因组的完整性和污染程度。
Prodigal 预测开放阅读框 (ORF),然后将它们翻译成为氨基酸序列。HMMER v3.1用于查找基因组中的31个单拷贝持家基因。分别使用数据库CARD v3.1.3和CAZy(202001更新)鉴定可能的抗生素耐药基因和碳水化合物活性酶,参数为e-value: 1e-5。VFDB 2022 数据库用于识别已确定病原体菌株的潜在毒力因子。
细菌鉴定策略如下:
1. 对 31 个单拷贝持家基因图谱进行 HMM 搜索后,从预测基因中提取单拷贝持家基因序列。
2. 对照 31 个单拷贝看家基因数据库,比对每个看家基因(参数e-value:1e-5),并将每个基因具有相同得分的前 200 个Blast结果保留。对于数据库中的每个物种,我们计算了Blast结果中包含该物种的持家基因的数量,并根据数量对物种进行排名。默认情况下,分析流程会过滤掉持家基因数量小于 15 的物种,但用户可以修改此阈值。因此,上述策略不仅可以识别培养的单一微生物,还可以识别污染样本。
3.计算样品基因组之间的ANI值,基因组来源于上述方法中选出的基因组,
仅报告一个物种的最大ANI值。对于一些含有过多菌株的物种,我们选择了多达 1000 个菌株进行 ANI 计算。
4. Barrnap v0.9 (https://github.com/tseemann/barrnap) 用于预测 16s rDNA。使用IQ-TREE v1.6.12构建了16s rDNA和持家基因的系统发育树。
此外,如果鉴定的物种被纳入PubMLST数据库(http://pubmlst.org),分析流程则会自动分析样品的分子分型。工作流程如图 2C 所示。
我们从NCBI的SRA数据库下载的样本数据,登录号为:SRR12560292。样本数据包含 1,418,820 个reads,产生了46 个scaffolds, 组装后的大小为 2.76Mbp。31个单拷贝持家基因富集于金黄色葡萄球菌,其中金黄色葡萄球菌S3是数据库中的结果最为接近。其MLST分型为ST22。共有142个基因被鉴定为在CARD数据库中注释为具有抗生素抗性,该样本中有462个VFs(图3)。
我们下载了 560 个 ATCC 标准菌株的基因组,以测试我们鉴定程序的准确性。只有5个基因组的鉴定结果与原菌株名称不一致。通过仔细分析,发现其中3种是由数据库中参考物种的命名错误引起的(GTDB数据库已根据WGS更正了其名称)。另外2个对代表性菌株的命名存在争议。MIST所有的鉴定都来自数据库中得分最高的基因组(表S2-S4)。
分析流程3:基于SNP的溯源分析
在食品药品质量控制过程中,除了微生物物种鉴定外,我们还需要分析某个物种的不同分离株之间的进化关系。例如,在制药厂环境中,我们可以通过分析分离株之间的进化距离来确定菌株污染的来源。
本系统集成了两种利用SNP系统发育进行微生物溯源的模式,分别通过EToki v1.2 和kSNP v3.0软件实现上述功能。在EToki模式下,通过将基因组与参考基因组进行比较找到SNP,并使用生成的一致性序列文件构建最大似然系统发育树。kSNP 是一种用于 SNP 鉴定和系统发育分析的程序,无需基因组比对或参考基因组,当相关微生物无法培养或种内进化距离较大时,kSNP更具优势。此外,两种模式都提供了系统发育树视图。工作流程如图 2B 所示。
扩增子数据库和参考基因组数据库
SILVA v138 和 UNITE v8.0作为扩增子参考数据库的来源,用于 16S rDNA/18S rDNA/ITS 微生物群落多样性分析流程的微生物鉴定。参考数据库的详细信息如表 1 所示。
此外,我们还建立了一个涵盖 223,491 个细菌 RefSeq基因组的持家基因数据库,以便在 WGS 工作流程中快速准确地分析微生物鉴定。从各基因组中提取31个单拷贝持家基因(dnaG、frr、infC、nusA、pgk、pyrG、rplA、rplB、rplC、rplD、rplE、rplF、rplK、rplL、rplM、rplN、rplP、rplS、rplT、rpmA、rpoB、rpsB、rpsC、rpsE、rpsI、rpsJ、rpsK、rpsM、rpsS、smpB和tsf),构建完整的数据库,共包含6,855,279个氨基酸序列。31个单拷贝持家基因数据库也用于基于全基因组测序技术的菌种鉴定。
结 论
全基因组测序、扩增子和宏基因组测序越来越多地用于产生复杂环境序列数据集的研究,这为不可培养微生物的鉴定和整个生物群落的利用打下了坚实的应用基础。因此,亟需在食品安全和药物管制领域开发基于WGS和扩增子的微生物物种鉴定流程。MIST系统可利用扩增子序列进行微生物鉴定,利用WGS数据可进行微生物鉴定、MLST分型和SNP源跟踪。在我们的系统中,WGS微生物鉴定流程的一个重要潜在用途是鉴定受污染的序列或宏基因组样本。现有分析方法鉴定环境微生物污染样本的能力十分有限,本系统在加快临床实验室病原体检测方面具有重要价值。
引文格式:
Song, Minghui, ChangHan, Linmeng Liu, Qiongqiong Li, Yiling Fan, HaoGao, Dan Zhang, Yi Ren, Feng Qin, and MeichengYang. 2023.“MIST: A Microbial Identification andSource Tracking System for Next‐GenerationSequencing Data.” iMeta e146.https://doi.org/10.1002/imt2.146
作者简介

宋明辉(第一作者)
● 上海市食品药品检验研究院首席青年研究员。
● 依托国家药监局药品微生物检测技术重点实验室,开展微生物鉴定溯源与风险识别技术创新研究。获评2022年上海市科技进步奖二等奖、2019年上海药学科技奖二等奖科研成果奖。承担和参与国家重点研发计划、药典会、上海市科委等课题30余项,发表学术论文30余篇,其中SCI论文13篇,申请国家专利12项、获软件著作3项,起草国家和团体标准9项。

韩畅(第一/通讯作者)
● 毕业于哈尔滨工业大学,上海美吉生物医药科技有限公司产品经理。
● 负责蛋白组学和转录组学的数据分析和挖掘与云平台产品的研发设计工作。相关成果取得国家发明专利1项,软件著作权1项。

刘林梦(第一作者)
● 上海交通大学生物学硕士。
● 目前研究方向是微生物组、微生物比较基因组以及多组学联合的数据分析与挖掘,数据库和软件研发经验丰富。部分成果发表于Nucleic Acids Research、The ISME Journal、iMeta等期刊。

任一(通讯作者)
● 上海美吉生物医药科技有限公司。博士毕业于南开大学。
● 自2002年起便从事基因组学研究,有20年基因组学、生物信息学研究经验。其领导开发的生物信息云计算平台为超过3,000+国内外知名研究单位和企业,60,000+科研工作者提供服务。发表SCI期刊论文23篇,包括PNAS、ISME、Nature communication、iMETA等。先后主持国家和市级科研项目6项,获8项授权发明专利。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 1126
1126

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









