






基于单细菌16S rRNA的菌群高通量精准识别与绝对定量方法BarBIQ
High-throughput identification and quantification of bacterial cells in the microbiota based on 16S rRNA sequencing with single-base accuracy using BarBIQ

Article,2023-11,Nature protocols,[IF 14.8]
DOI:10.1038/s41596-023-00906-8
原文链接:https://www.nature.com/articles/s41596-023-00906-8
第一作者:Jianshi Jin (金坚石)
通讯作者:Jianshi Jin (金坚石);Katsuyuki Shiroguch
合作作者:Reiko Yamamoto
主要单位:
中国科学院动物研究所 (State Key Laboratory of Integrated Management of Pest Insects and Rodents, Institute of Zoology, Chinese Academy of Sciences, Beijing, P. R. China)
日本理化学研究所(Laboratory for Prediction of Cell Systems Dynamics, RIKEN Center for Biosystems Dynamics Research (BDR), Osaka, Japan)
- 亮点 -
• 详细描述了一种鉴定及定量菌群样品中细菌的新方法——BarBIQ (Barcoding Bacteria for Identification and Quantification)
•BarBIQ使用细胞条形码及基于液滴的一步扩增策略得到了相比传统16S rRNA测序方法更精确的细菌物种鉴定和定量结果
- 摘要 -
细菌通常以不同细菌组成的群落即菌群为单位发挥作用。准确地鉴定一个群落中的细菌同时定量每种细菌的数量可以增进我们对菌群的认识。然而,目前传统的16S rRNA基因测序方法只能识别与定量基因,而基因与细胞并不对等,因此上述目的不能通过传统方法完成。本文介绍我们开发的方法即BarBIQ(Barcoding Bacteria for Identification and Quantification)的详细流程。在BarBIQ中,单个细菌的16S rRNA基因在一个液滴中被扩增并连接上序列唯一的细胞条形码。对来自不同液滴的串联连接的细胞条形码与16S rRNA基因进行测序后,通过细胞条形码区分每个单细菌,根据 16S rRNA 基因确定每个单细菌的种类,并根据条形码数量定量每种细菌的细胞数量。BarBIQ通过使用液滴避免不同细菌的16S rRNA基因形成嵌合体实现了16S rRNA基因测序的单碱基精度。对于数据处理,我们开发了一个易于使用的生物信息学分析流程 (https://github.com/Shiroguchi-Lab/BarBIQ_Pipeline_V1_2_0)。对于具有分子生物学知识但没有生物信息学背景的研究者,完成该方法流程需要约2周时间。本文以BarBIQ在小鼠肠道菌群分析中的应用为示范。同时,该方法也适用于其他菌群样品,包括来自口腔和皮肤、海洋、土壤和植物的样品以及来自其他陆地环境的样品。
- 引言 -
在海洋或陆地环境中,许多不同的种类细菌通常会聚集成群落与其他生物(如人、动物、植物)共生。微生物-宿主互作可以在宿主内稳态的建立中发挥重要作用,因此,研究这些菌群的组成是很重要的。为此,我们需要一种高通量方法来鉴定并定量菌群中的细菌。传统的高通量16S rRNA基因扩增子测序方法(如OTU及ASV法)只能鉴定16S rRNA基因类型并量化每种基因类型的扩增数量。然而不同的细菌的基因组中有着不同的16S rRNA基因拷贝数,即使相同的细菌也可能有不同的16S rRNA基因类型。因此,使用传统方法不能根据测序的结果完全鉴定单细菌的类型,同时也不能定量菌群中每种细菌的数量。
- 方法开发 -
为了克服传统16S rRNA基因测序方法的局限,我们开发了BarBIQ方法,这是一种对菌群中单个细菌进行高通量鉴定和定量的方法,该方法采用条形码标记单细菌的策略并实现了对16S rRNA基因鉴定的单碱基精确度。BarBIQ与传统的16S rRNA测序方法有本质区别:首先,BarBIQ可以鉴定每个细菌的种类,并且可以定量每种细菌的细胞数量,而传统方法只能鉴定16S rRNA基因序列并量化每种基因序列的扩增数量。第二,BarBIQ可以参照测量的相对细胞数与总的细胞数的结果对菌群中每种细菌进行绝对定量,而传统方法是基于基因扩增数量的比值结合测量的总细胞数量对OTU进行绝对定量的。为使研究人员清楚了解这种基于细胞的新概念,我们引入了一个新的计量单位cOTU(cell-based OTU),即用基于细胞的16S rRNA序列类型及细胞数量来描述细菌菌群。第三,BarBIQ对16S rRNA基因的鉴定具有单碱基准确性与分辨率,而传统方法对16S rRNA基因的鉴定会出现错误。对于同样的生物学问题,不同的定量方法可能得到不同的结论,如基于细胞还是基于分子或者基于绝对定量还是相对定量。
BarBIQ的实验方案与传统方法相比有很多的新颖之处。例如,对于样品处理, 我们开发了一种可以测量每毫克样品中有多少个细胞的方法,同时建立了一种可以分离细胞与细胞外DNA的方法。对于文库构建,我们开发了一种利用液滴将细胞条形码与单一细菌的16S rRNA基因连接的方法:首先,我们设计了一套可以在一次 MiSeq测序中区分10万个以上细菌细胞的细胞条形码;其次,我们开发了一种通过涡旋将聚集的细菌团块打散,并基于泊松分布调整条形码和细菌的浓度及其比例以使每个液滴中只有一个细菌细胞的方法;第三,为了避免不同细菌在扩增过程中产生嵌合体,我们开发了一种在每个液滴内一步生成文库的方法,这个过程包括通过加热裂解细胞、条形码与16S rRNA基因同时扩增、连接测序接头以及将扩增的条形码与细菌16S rRNA基因连接等。
对于Illumina MiSeq测序,我们设计了特定的spike-in文库来保持序列的异质性从而最大限度的提高测序质量。我们还开发了一个独特的序列读取系统。我们用30个循环测序细胞条形码(Illumina平台上的“R1”),295×2个循环测序16S rRNA基因(分别为“I1”和“R2”),8个循环测序样品标签(“I2”)。此外,我们用定制引物替换了Illumina Index 1测序引物。
对于数据分析,我们开发了一个自动化的分析流程(https://github.com/Shiroguchi-Lab/BarBIQ_Pipeline_V1_2_0),其中包括以下过程:基于细胞条形码和16S rRNA序列的聚类、16S rRNA基因序列错误的纠正、同一个细菌中不同类型16S rRNA基因的识别、已鉴定的每种细菌的数量的计算、实验环境污染的识别。每一步的具体算法请参考Jin et al, Nat. Commun., 2022, doi: 10.1038/s41467-022-28426-1。
- 方法流程概述 -
该方法具体流程包含以下步骤(图1)。首先,将收集的菌群样品混匀到缓冲液中,通过涡旋使聚集的细菌团块分开,过滤离心分离细菌完整细胞和细胞外的DNA,然后用微滴数字PCR测量每毫克样品中的细菌总细胞数。接着在测定细胞条形码混合液的浓度后,将细菌样品与细胞条形码、引物和试剂混合,并将混合液制成液滴(直径:~ 120 µm)用于DNA扩增。基于泊松分布调整条形码和细菌的浓度及其比例,确保90%以上同时含有细胞条形码和细菌的液滴仅有一个细菌细胞(对于细胞外DNA,基于泊松分布调整条形码和含有16S rRNA基因的DNA片段的浓度及其比例,以确保90%以上同时含有细胞条形码和DNA片段的液滴仅有一个DNA片段)。之后,每个液滴中的16S rRNA基因和条形码被扩增并串联连接,同时将连接产物连接上测序接头片段。接下来将来自所有液滴的文库(连接的扩增子)回收,并通过AMPure XP beads进行两轮纯化、两轮凝胶纯化,再使用链霉亲和素包被磁珠进行两轮纯化用以在浓缩前去除生物素标记的被扩增但未连接细胞条形码的16S rRNA基因。接着用qPCR对文库进行质检和定量。然后,把扩增文库与制备并纯化的spike-in文库混合后,使用MiSeq测序仪对扩增后的16S rRNA基因以及细胞条形码进行测序。最后,分析每个细胞条形码的测序结果来识别其对应的菌种并计算每种菌的数量。通过使用测量过的每毫克样品包含的细胞总数量计算出每种细菌的绝对数量(对于细胞外DNA,通过测量的每毫克的总DNA量来进行绝对定量)。值得注意的是,因为相同的细胞条形码会与来自相同细胞的多个扩增的16S rRNA序列相连,所以这种分析适用于基因组中具有多种16S rRNA序列类型的细菌。
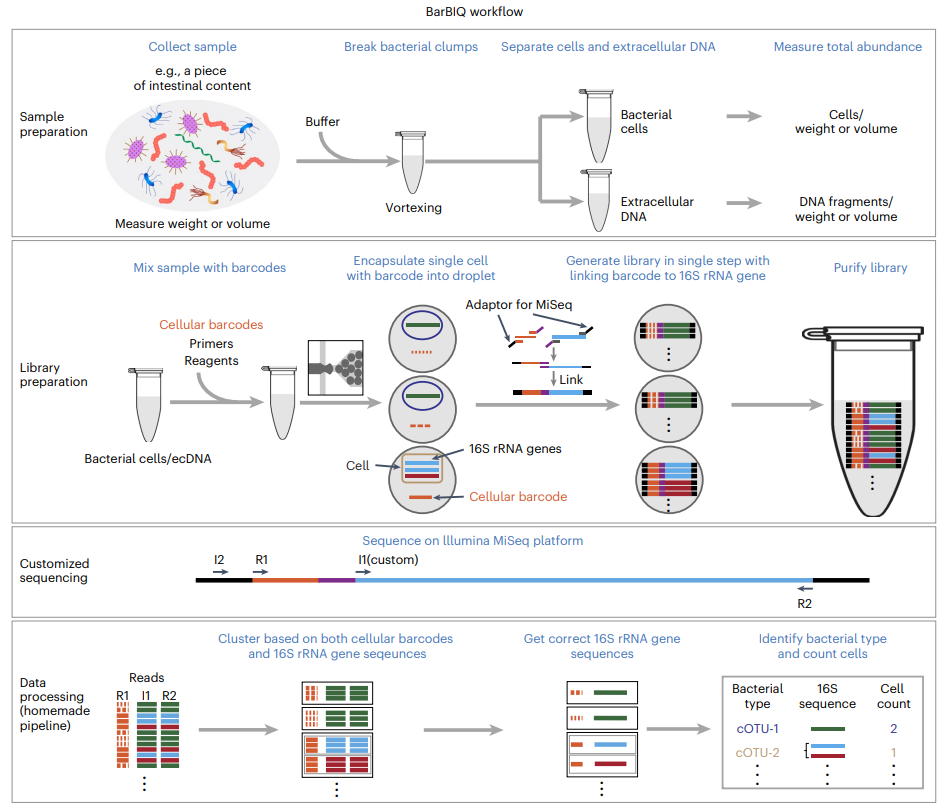
图1 BarBIQ工作流程图。
- 应用方向 -
作为示范,在我们之前的研究中,BarBIQ已被应用于包含10种可培养且具有已知16S rRNA基因序列和细胞丰度的人类肠道细菌菌株的模拟菌群。应用BarBIQ,10种细菌均被正确鉴定,每个菌种的细胞数量也被准确量化。在同一项研究中,BarBIQ被应用于雄性C57BL/6J小鼠的盲肠菌群(22个来自11只小鼠盲肠远端和近端位置的样品),该研究能够检测到菌群受饮食中维生素A的细微影响,发现了小鼠盲肠远端位置大约200个被检测到的cOTU中只有一个cOTU的细胞数量会发生变化。在小鼠盲肠中,BarBIQ鉴定出了383个新的未在三个广泛使用的公共数据库(GreenGenes、Ribosomal Database Project以及Silva)中注册的16S rRNA序列(占所有已鉴定序列的38%)。
除了上述示例以外,BarBIQ还可应用于其他来源的菌群样品,对其进行每种细菌的鉴定与定量分析,如来自口腔、皮肤、海洋、土壤和植物的样品以及来自其他陆地环境的样品。此外,BarBIQ还可以提供一个新的数据库,其中包含已鉴定类型的细菌的信息、每种细菌类型的数量,以及准确识别的16S rRNA基因序列。基于细胞的定量分析可以与诸如组学分析和成像等其他分析方法结合。最后,菌群中细菌的量化可用于研究宿主菌群与动植物疾病之间的相关性或监测受环境因素影响的变化。
- 方法比较 -
最近,一些关于细菌菌群的单细胞基因组测序方法被陆续报道。尽管这些方法在细菌分类的分辨率和功能基因的分析上优于16S rRNA基因扩增子测序方法,但由于测基因组比测单基因需要更多的测序资源,其分析通量远远低于16S rRNA测序方法。具体来讲,细菌的基因组大小是16S rRNA基因大小的约1000倍,因此一次实验中可以测量细胞的通量单细胞基因组测序应该比16S rRNA测序方法BarBIQ低大约一千倍。此外,因为单细胞基因组测序方法对基因扩增的效率较低,会造成不同细胞扩增的不均一,所以这些方法定量每种细菌细胞数量的准确性低于BarBIQ。
- 优势与不足 -
BarBIQ的关键优势在于其能够在识别细菌类型的同时定量每种细菌的细胞数量,它能够同时表征菌群整体和菌群中的单个细菌,为进一步了解细菌-宿主相互作用机制打下基础。BarBIQ还可以通过共同的表征单位——细胞(或每个细胞)直接与例如代谢组分析和空间分析的其他分析方法相结合使用。此外,序列的无错误鉴定使得BarBIQ可以鉴定新的16S rRNA序列和新的细菌,可供广泛使用的公共数据库扩展新的正确的数据。
因为BarBIQ以16S rRNA基因的一部分(大约500个碱基)为靶点来鉴定细菌,所以如果某些细菌的16S rRNA基因在该区域中没有差异则无法对它们有效地区分。当然,传统的方法也存在这种问题。对于这个问题,可以为BarBIQ设计适当的引物并使用长读长测序仪实现对全长16S rRNA基因的测序甚至是更长的16S-ITS-23S rRNA操纵子的测序来解决。此外,为BarBIQ设计合适的引物,还可用于鉴定其他DNA序列,如古细菌的16S rRNA基因、真菌的18S rRNA基因、病毒序列以及哺乳动物细胞中的特定基因等。
- 实验设计 -
该方法适用于任何即时取得或细菌细胞尚未破碎的样本量足够的菌群样品。例如,本方法流程可直接应用与人类或其他哺乳动物的大肠内容物或粪便样品。该流程设计每个细胞样本和细胞外DNA样本一次测量24万个细胞或分子,最终大约2万个细胞或分子可以被检测到。需要注意的是,因为不同的样品可能具有不同的细菌密度,所以测量所需的样品总重量取决于样品的类型。因此,我们建议在进行一个大样本量的实验之前,先通过步骤9-16(见原文详细实验流程,下同)来确定样品的细菌细胞数量及细胞外DNA分子数量以优化实验方案。此外,一些菌群样品(如土壤样品)中的杂质颗粒可能会堵塞步骤13中的液滴生成器(DG8 Cartridges),因此优化实验方案防止堵塞的发生也是必要的。
为了获得合适的测序深度,一次MiSeq测序可以对6个菌群样品中获得的6个细胞样本库和6个细胞外DNA样本库以及这些样本的实验环境对照文库同时进行测序。请注意,实验环境对照数量取决于具体的实验设计,比如对6个菌群样本的对照数量可以在2-12之间调整,具体请参阅步骤3。因为测序超过500个碱基才能覆盖16S rRNA基因的V3-V4区域,所以我们建议使用MiSeq;但如果允许使用较低分辨率对细菌类型进行分类,可以使用短读长测序仪来运行低于295×2个循环的测序步骤(对应Illumina平台的“I1”和“R2”) (请参考原文步骤174-185)。
如需测序细菌16S rRNA基因的其他区域,或者古细菌和真菌等其他生物,可以将引物P7-R2P-341F中的“CCTACGGGNGGCWGCAG”序列和引物Biotin-
Link-805R中的“GAC/iBiodT/ACHVGGGTATCTAATCC”序列替换为所需扩增目标新设计的引物。
详细实验和分析流程请参阅原文。
- 预期结果 -
执行该方法流程后将得到以下结果:一个包含来自所有样品中被检测到的16S rRNA序列的V3-V4区域(被标记为Bar sequences)及cOTU信息的excel文件;一个包含Bar sequences及其cOTU ID和RDP classifier预测得到的门到属的分类信息的fasta文件;以及一个包含每个样品中检测到的每个cOTU的细胞数(或DNA片段数)、单位重量或体积内的细胞(或DNA片段)的绝对数量、相对丰度及RDP classifier预测得到的门到属的分类信息的的excel文件。
作为示例,我们使用本方法流程检测了包含10种培养的具有已知的16S rRNA基因序列和丰度的人类肠道细菌菌株的模拟菌群以及来自C57BL/6J雄性小鼠远近两段的盲肠内容物,并获得了以下代表性结果。BarBIQ从模拟菌群中正确鉴定出了10种细菌,其中包含了在3个技术重复中相同的16条16S rRNA基因序列。此外我们还测定了模拟菌群中每个cOTU单位体积内的细胞数,它们在3个技术重复中表现一致(标准差/平均值:0.01 ~ 0.25,N = 3;图2)。
BarBIQ鉴定并定量了小鼠盲肠细胞样本中的细菌细胞和小鼠盲肠内的细胞外DNA中的16S rRNA基因。共计测到3.4 105的细菌细胞并鉴定了810个包含954条16S rRNA基因序列的cOTU。我们还从细胞外DNA样品中鉴定到了另外的50条16S rRNA基因序列。使用RDP classifier,对每个cOTU中的16S rRNA基因序列进行了从门到属水平的分类预测,最终将810个cOTU分类到7个门,13个纲,21个目,42个科和103个属。对于细胞样品,我们量化了已鉴定cOTU单位重量中细胞的绝对数量;对于胞外DNA样品,我们同样对已鉴定的cOTU或Bar序列单位重量中含有16S rRNA基因的DNA片段的绝对数量进行了量化。对于每个样本中每个cOTU的定量分析,首先,通过对相同样本的技术重复上的比较发现,该方法获得的结果具有高度可重复性(图2b)。其次,通过对每毫克样本包含的总细菌数量的测定,最终计算出每种cOTU的绝对数量。
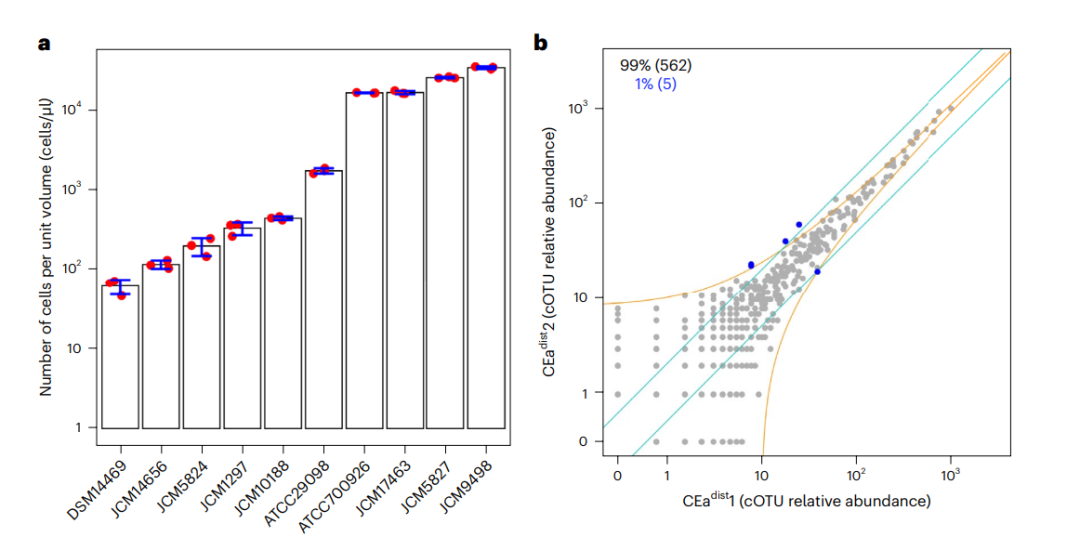
图2 :模拟菌群和小鼠盲肠菌群的BarBIQ定量结果。
a.由BarBIQ测得的模拟菌群中每个菌株(x轴)单位体积的细胞数。
b.比较BarBIQ对相同样本的两个技术重复测定的每个cOTU(BarBIQ鉴定的细菌类型)的相对丰度。BarBIQ是通过测定每毫克样本包含的总细菌数量并乘以每个cOTU的相对丰度获得每个cOTU的绝对丰度。
该图表明BarBIQ具有很高的可重复性。
参考文献
Jianshi Jin, Reiko Yamamoto, Tadashi Takeuchi, Guangwei Cui, Eiji Miyauchi, Nozomi Hojo, Koichi Ikuta, Hiroshi Ohno & Katsuyuki Shiroguchi. (2022). High-throughput identification and quantification of single bacterial cells in the microbiota. Nature Communications, doi: 10.1038/s41467-022-28426-1
Jianshi Jin, Reiko Yamamoto & Katsuyuki Shiroguchi. (2023). High-throughput identification and quantification of bacterial cells in the microbiota based on 16S rRNA sequencing with single-base accuracy using BarBIQ. Nature Protocols, doi: 10.1038/s41596-023-00906-8
- 金坚石课题组招聘启事 -
中国科学院动物研究所-农业虫害鼠害综合治理研究国家重点实验室-菌群生物学与智能调控研究组,现因科研工作需要,拟招聘1名助理研究员和2~3名名博士后。欢迎具有生物实验技术、生物信息学、微生物学、动物学、免疫学、分子生物学、生物物理学或计算机等相关专业的博士申请。同时非常欢迎联合培养的研究生来课题组进行合作研究,有意向报考研究生和博士生的同学来研究组深入了解。
应聘成功的博士后,课题组将大力支持申请国家博士后创新人才支持计划、博士后国(境)外交流项目等。还可申报中国科学院特别研究助理资助计划、动物研究所‘秉志’博士后特别支持计划。出站的优秀博士后,可继续留组并晋升为助理研究员,特别优秀的可直接破格晋升为副研究员(均为中科院编制)。
有意者请将个人简历和应聘信通过E-mail提交(金坚石 E-mail: jinjs@ioz.ac.cn),邮件请命名为“应聘岗位+姓名+研究方向”。
课题组简介
菌群在生态系统中处于非常重要的地位,不但参与环境中能量和物质的循环,同时也对宿主(如动物)的健康和疾病至关重要。这为利用菌群预测或控制农业有害动物爆发、保护生物多样性、改善人类健康和生活质量等提供了新思路。本课题组将通过研发和应用基于大数据采集与分析的精确技术,特别是基于测序和成像的单细胞技术,从环境或共生菌群发现重要的功能微生物并研究其生物学,在此基础上结合人工智能和生物技术实现对菌群的精准调控,同时为调控动物的繁殖与行为、改善环境或宿主健康等提出理论或开发具体技术。详情请访问https://www.jinlab.ac.cn/
课题组长简介

中国科学院动物研究所
金坚石
研究员
金坚石,博士,研究员,博士生导师。2009年和2014年分别获得南京大学学士和北京大学博士学位。2010-2011前往哈佛大学访问学习。2014-2016年在北京大学生命科学学院从事博士后研究。2016-2023年在日本理化学研究所先后担任JSPS访问研究员、Research Scientist、Senior Scientist。2023年3月回国担任中国科学院动物研究所研究员,现已获得国家重点研发计划课题等多项国家级课题,经费充足。
主要从事单细胞测序、显微成像和智能化系统等的研发工作,研究肠道菌群和转录调控等复杂系统相关的生物学问题。研究成果发表于Science,Nature Communications, Nature Protocols, Science Immunology,PNAS,The Journal of Immunology等杂志。已申请多国PCT专利。曾获得日本理化学研究所“樱舞赏”,日本JSPS博士后奖学金,北京大学优秀博士论文,北大-清华生命科学联合中心优秀博士后基金等。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









