








整合16.8万个样本揭示人类肠道微生物组的全球模式
● 期刊:Cell (IF:45.5)
● DOI:https://doi.org/10.1016/j.cell.2024.12.017
●原文链接: https://www.cell.com/cell/fulltext/S0092-8674(24)01430-2
● 第一作者:Richard J. Abdill & Samantha P. Graham
● 通讯作者:Ran Blekhman(blekhman@uchicago.edu)
● 发表日期:2025-1-22
● 主要单位:
美国芝加哥大学医学部遗传医学、美国明尼苏达大学遗传学、细胞生物学与发展系、美国科罗拉多大学医学院生物医学信息学系、美国科罗拉多大学医学院健康人工智能中心、美国科罗拉多大学安舒茨医学园区公共卫生学院生物统计学与信息学系、美国加州大学圣地亚哥分校医学院儿科系
亮点Highlight
● 在 microbiomap.org 上处理和整合了 168,000 份公共 16S 肠道微生物组样本
● 不同地区的微生物组组成和多样性差异很大
● 扩增子选择等技术因素与组成差异有关
● 根据简编数据训练的分类器可从组成推断其所在世界地区
摘要Summary
影响人类微生物组变异的因素是生物医学研究的一个主要焦点。尽管其他领域已经通过利用大规模测序数据集获得了需要庞大样本量才能得出的见解,微生物组领域却一直缺乏一个规模相当的资源,以支持常用的16S rRNA基因扩增子测序来量化微生物组组成。为了填补这一空白,我们采用统一的方法处理了 16,8464 份公开的人类肠道微生物组样本。我们利用这份数据集来评估地理和技术对微生物组变异的影响。我们发现,中亚和南亚等地区的微生物组与欧洲和北美洲那些已得到充分研究的微生物组有很大不同,而且仅凭微生物组的组成就能预测样本的来源地区。我们还发现微生物群变异与引物使用、DNA 提取等技术因素之间存在密切联系。我们预计,人类微生物组综合数据库这项不断发展的工作将促进先进的应用和方法研究。
引言Introduction
人类微生物组是理解健康与疾病的一个重要因素。在健康个体与患有与微生物群相关疾病(如结直肠癌、炎症性肠病)的个体之间,已观察到微生物组组成的系统性差异,这强调了理解微生物组变异决定因素的重要性。研究表明,这种变异受到宿主遗传因素和种族等因素的驱动,其中许多因素与地理区域相关。例如,不同国家之间的饮食纤维和加工食品的消费量存在差异,而抗生素的使用情况也是如此,这两者都已知会影响肠道微生物群。微生物组组成将地理位置、文化和人类健康联系起来,这一动态在从泰国、拉丁美洲和韩国移民到美国个体所经历的组成变化中得到了体现。
尽管理解世界各地区、文化和社交群体之间的微生物组变异至关重要,但许多人群实际上被排除在文献之外。在我们之前的研究中,我们发现高收入国家(如美国)在公共数据库中被过度代表,而其他地区(如东亚国家)则相对采样不足。与其他领域一样,许多公开可用的微生物组数据集可以用于量化最广泛研究的世界地区与那些相对未被充分研究的世界地区之间的差异。
然而,这种分析受到众多技术因素的影响,这些因素可能会改变观察到的微生物组组成,包括样本收集、保存和存储、测序平台以及提取的DNA浓度。尤其是提取试剂盒的选择受到了特别关注,特别是其中包含的珠磨步骤,该步骤通过一组小珠子对细菌细胞进行震荡破碎的机械法,对传统的酶解法形成补充或替代,从而获取DNA。许多研究发现,这种机械裂解步骤可以显著增加观察到的分类单元数量以及相对丰度。
在人类肠道这样复杂的环境中,只有在收集了数千个样本后,重要的模式才可能变得明显。像recount3这样的大型综合数据集已经在转录组分析中揭示了复杂微生物基因表达模式的菌株水平差异,以及可用于增强转录组关联研究的人类基因表达模块。为了填补微生物组领域缺乏类似规模资源的空白,我们在此介绍了人类微生物组综合数据库,这是一个包含超过168,000个公开可用的人类肠道微生物组样本的集合,这些样本来自68个国家。所有样本都经过统一的流程重新处理,并合并成一个单一的数据集,该数据集以多种格式在本文和microbiomap.org上提供。我们通过评估全球范围内的肠道微生物组组成模式,量化了我们对人类肠道微生物组当前知识的差距,并识别了常见技术因素对微生物组量化的影响,从而展示了该综合数据库的价值。
结果Result
统一处理实现了大规模全球微生物组多样性的量化
为了生成人类微生物组综合数据库(Human Microbiome Compendium),我们从美国国家生物技术信息中心(NCBI)的生物样本数据库(BioSample database)中识别出245,627个16S rRNA基因扩增子测序样本。这些样本被组织成研究项目(“BioProjects”)。我们专注于基于Illumina的检测方法,并排除了报告使用焦磷酸测序(pyrosequencing)和长读长测序数据的BioProjects。我们使用“Divisive Amplicon Denoising Algorithm 2”(DADA2)为每个BioProject生成一个分类表,其中每一行是一个样本,每一列是一个单独的分类单元(taxon)。为了在BioProjects之间整合数据,我们处理并量化了每个BioProject的扩增子序列变异(ASVs),每个ASV代表该BioProject样本中观察到的一个独特序列。
随后我们使用相同的参考数据库(本研究中为SILVA v138.0;见STAR Methods部分,了解数据集中可用的其他分类)将每个ASV尽可能具体地分类到属水平。最终结果量化了每个样本中分配给每个分类单元的读数数量。我们对482个BioProjects重复了这一过程,最终生成了一个包含168,464个样本的综合数据库,涵盖68个国家,涉及5.57万亿碱基的测序数据,所有样本均使用统一的流程进行处理(图1A)。有关数据处理流程和质量控制的详细描述,请参阅STAR Methods部分。我们还开发了自动注释流程,以推断样本的来源国家以及技术因素(如提取试剂盒和扩增子选择)。结合对出版信息和研究人群的手动整理,我们能够在全球范围内量化肠道微生物组组成的模式,并使用户能够将这些经过统一处理的数据进一步细分,以更贴合他们自己的研究问题。经过处理的数据—人类微生物组综合数据库—可在微生物组地图网站(microbiomap.org)上免费以多种格式获取(见资源可用性部分),用户可以在该网站上浏览、可视化、筛选和下载数据及元数据。我们还创建了一个R软件包MicroBioMap,以便进一步分析这些数据。我们相信,这个大型综合数据库非常适合与微生物生态学应用相关的日益丰富的机器学习工具,这些工具在GMrepo和mBodyMap等项目之后,为统一处理的16S人类肠道样本提供了更广泛的访问权限(表S1)。
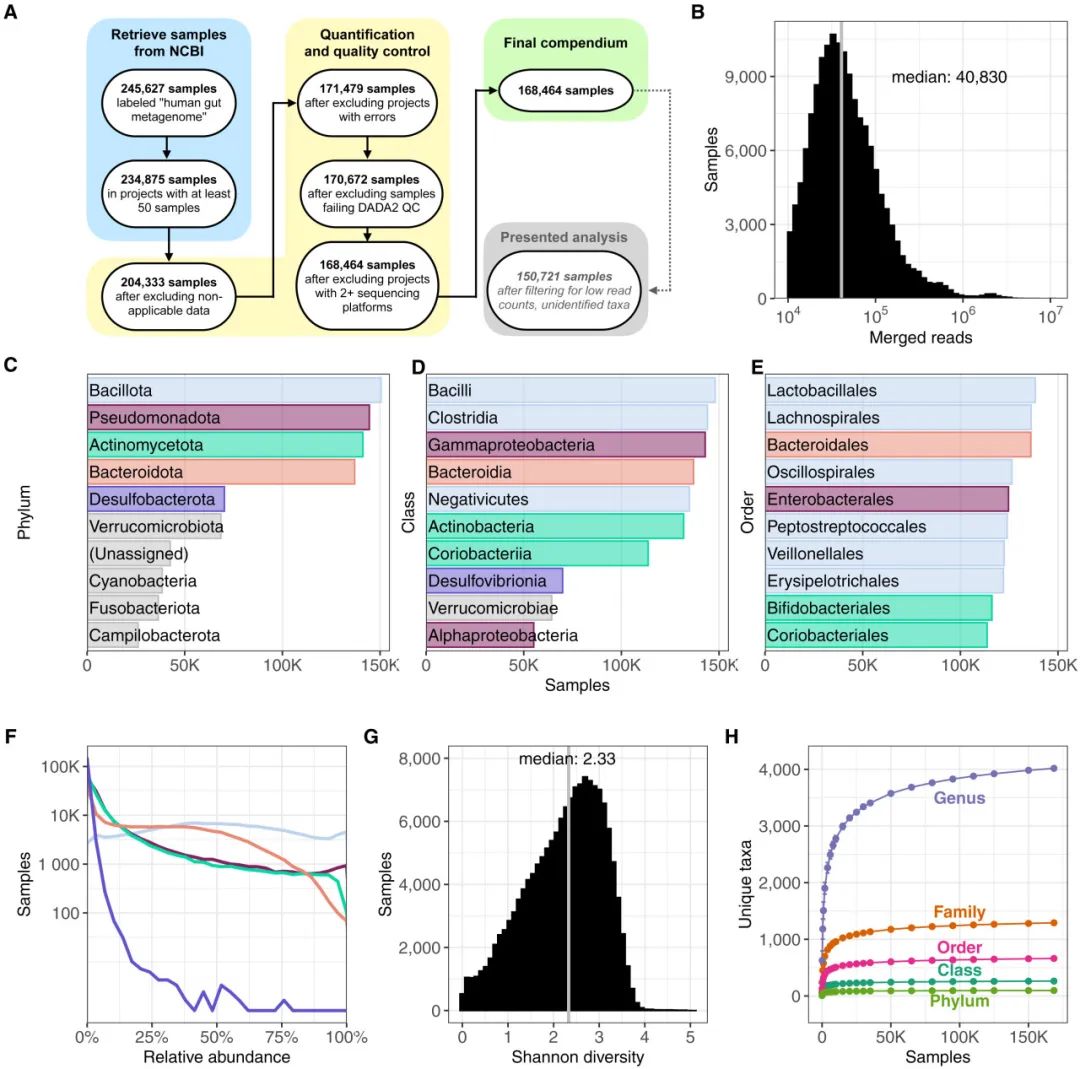
图 1 | 人类微生物组综合数据库概览
(A)图中列出了数据处理的一般步骤以及每个步骤完成的样本数量。详细步骤见STAR Methods部分。
(B)直方图展示了每个样本中分类的读取量分布。x轴表示每个样本的读取量,y轴表示具有该读取量的样本数量。灰色垂直线表示中位数读取量为40,830。
(C–E)展示了综合数据库中观察到的最常见分类单元。每个样本中的读取量被分配到尽可能具体的分类单元名称,直到属水平。每个面板展示了在三个最高分类水平(门、纲、目)中整合这些分配的结果。(C)显示了最常见门水平分类单元,前五名分别用不同颜色表示。这些颜色在后续两个面板中用于指示每个分类单元所属的门。(D)显示了数据集中观察到的最常见细菌纲。(E)显示了数据集中观察到的最常见目。
(F)密度图展示了数据集中门水平相对丰度的分布。每条线代表数据集中最常见门之一,使用与图C相同的颜色。灰色线表示其他所有门。x轴表示单个样本中给定门的相对丰度,y轴(伪对数尺度)表示观察到该分类单元丰度的样本数量。
(G)直方图展示了综合数据库中观察到的Shannon多样性分布。x轴表示样本的α多样性(使用Shannon多样性指数计算),y轴表示观察到该得分的样本数量。请注意,这些样本未进行稀释,该图反映了样本中不同读取量的多样性。
(H)稀释分析图展示了不同规模模拟综合数据库中观察到的独特分类单元数量。x轴表示综合数据库中的微生物组样本数量,y轴表示在重复抽样中观察到的平均独特分类单元数量。每条线表示在不同分类水平上的独特分类单元数量。
为了进一步分析,我们创建了一个过滤后的综合数据库,包含150,721个样本,每个样本至少有10,000个读数(见STAR Methods部分),以排除低质量样本和过于罕见而无法在BioProjects或世界地区之间进行比较的微生物。中位样本包含40,830个读数,经过修剪、质量过滤和配对读数合并后,9.6%的样本包含超过150,000个读数(图1B)。正如在人类肠道微生物组的早期测序研究中观察到的那样,我们发现厚壁菌门(Bacillota,原名“Firmicutes”)是最普遍的门(图1C),在150,721个样本中的150,540个(99.9%)中被发现,其次是假单胞菌门(Pseudomonadota,原名“Proteobacteria”;144,489个样本;95.9%)、放线菌门(Actinomycetota,原名“Actinobacteria”;93.7%)和拟杆菌门(B
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









