






点击蓝字 关注我们
利用HTBGC-Finder系统表征人类肠道微生物群中水平转移的生物合成基因簇

iMetaOmics主页:http://www.imeta.science/imetaomics/
研究论文
● 原文链接DOI: https://doi.org/10.1002/imo2.62
● 2025年2月1日,山东大学张磊、赵国屏院士团队在iMetaOmics在线发表了题为“Systematic Characterization of Horizontally Transferred Biosynthetic Gene Clusters in the Human Gut Microbiota Using HTBGC-Finder”的文章。
● 本研究开发了一种新型工具——水平转移的生物合成基因簇的搜索器(HTBGC-Finder),利用宏基因组数据,系统地识别了人类肠道微生物群中潜在的水平转移生物合成基因簇,为了解生物合成基因簇在肠道微生物群中的水平转移动态及其对宿主-微生物群相互作用的潜在影响提供了重要的见解。
● 第一作者:吴嘉诚、杨潇
● 通讯作者:张磊(zhanglei7@sdu.edu.cn)、赵国屏(gpzhao@sibs.ac.cn)
● 合作作者:赵兰兰、李紫云
● 主要单位:山东大学公共卫生学院、山东大学微生物国家重点实验室、中国科学院上海营养与健康研究所
亮 点
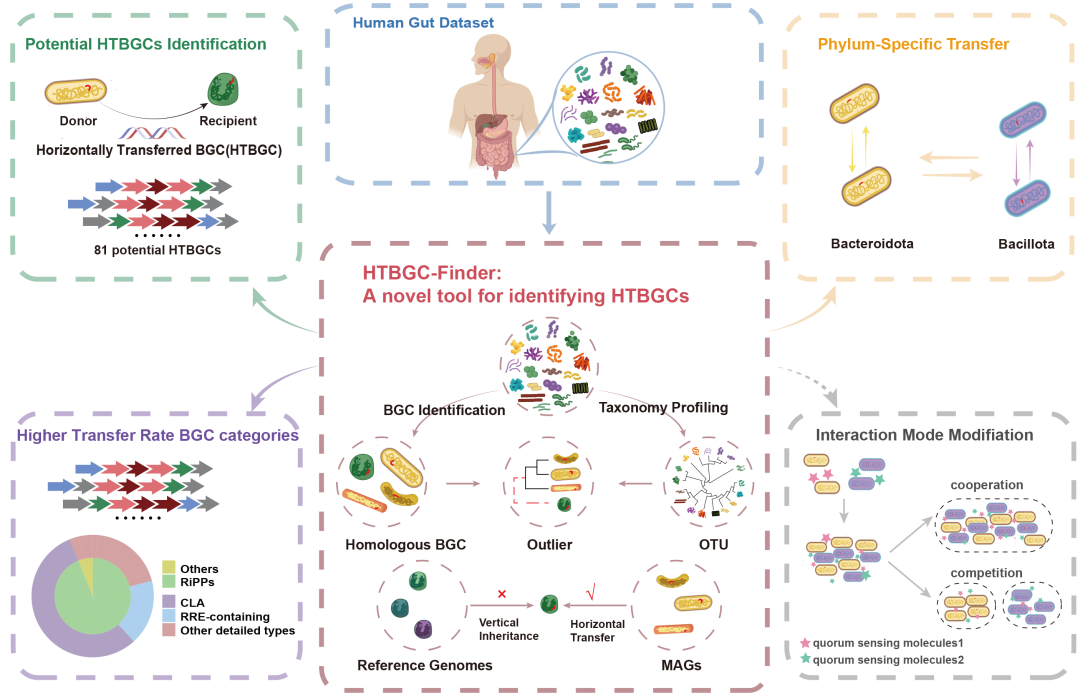
● 开发HTBGC-Finder:开发了一个新的生物信息学工具——水平转移生物合成基因簇的搜索器(HTBGC-Finder),用于识别潜在的水平转移生物合成基因簇;
● 鉴定水平转移的生物合成基因簇:利用 HTBGC-Finder 在人类肠道微生物群中发现了81个潜在的水平转移生物合成基因簇;
● 转移率更高:与非核糖体合成和翻译后修饰的肽(non-RiPPs)和非环内酯诱导体(non-CLA)相比,RiPPs和CLA的转移率明显更高;
● 特定门类的转移:只在Bacteroidota (Bacteroidetes)和Bacillota (Firmicutes) 内检测到生物合成基因簇的水平转移;
● 跨门类转移:观察到跨门类水平转移的实例,表明肠道微生物群内部可能存在快速进化的机制。
摘 要
人类肠道微生物群含有编码生物活性次级代谢产物的生物合成基因簇(BGC),它们在微生物与微生物以及宿主与微生物之间的相互作用中发挥着关键作用,是药物先导化合物的丰富来源。了解BGC的水平转移可以揭示微生物的适应性、资源利用和进化机制,从而推动生物技术的应用。尽管BGC在肠道微生物群中的水平转移非常重要,但人们对它的了解仍然很少。本研究介绍了一种新型工具——水平转移生物合成基因簇搜索器(HTBGC-Finder),旨在系统地识别广泛的人类肠道微生物群中潜在的水平转移BGC(HTBGC)。利用HTBGC-Finder,研究发现了81个潜在的HTBGC,这凸显了水平基因转移在塑造肠道微生物群方面的普遍性和重要性。值得注意的是,核糖体合成和翻译后修饰的肽(RiPPs)构成了这些HTBGC的大部分(81个中的76个,93.83%),与非RiPPs相比,其转移率明显更高(卡方检验,p < 0.001)。进一步研究发现,含环内酯诱导体(CLA)和RiPP识别元件(RRE)的BGCs几乎占RiPPs总数的四分之三(分别为45个即55.56%和14个即17.28%)。值得注意的是,CLA的转移率也高于非CLA(卡方检验,p < 0.001)。分类学分析表明,BGC水平转移只发生在类杆菌(Bacteroidota,亦称Bacteroidetes)和芽孢杆菌(Bacillota,亦称Firmicutes),分别为50和31例。此外,还观察到了跨门类的水平转移事件,凸显了肠道微生物群与宿主健康之间复杂的相互作用。这些发现为了解BGC在肠道微生物群中的水平转移动态及其对宿主—微生物群相互作用的潜在影响提供了宝贵的见解。
视频解读
Bilibili:https://www.bilibili.com/video/BV1mUArewEyV/
Youtube:https://youtu.be/4H9jbSaEKJA
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/imetaomics/
全文解读
引 言
人类胃肠道是一个重要的生态位,蕴藏着惊人的100万亿个微生物。这些肠道微生物总共拥330多万个基因,大约是人类基因组中基因总数的100倍。这种巨大的基因多样性使肠道微生物群被称为人体的“第二基因组”。肠道微生物群、人类基因组和环境因素之间的动态相互作用对人类健康产生了多方面的影响。
细菌和其他肠道微生物群的基因组中蕴藏着生物合成基因簇(BGC),由DNA片段组成,编码合成功能物质或执行特定功能。根据合成途径和编码产物的类型,BGC可分为聚酮合酶(PKS)、非核糖体肽合成酶(NRPS)、核糖体合成和翻译后修饰肽(RiPPs)——其中一些含有RiPP前体识别元件(RRE)、萜类、糖类和生物碱等。在微生物生长和发育过程中,BGC可协调产生各种次级代谢产物。这些物质在微生物的生态学和生理学中发挥着重要的作用,同时也是各种医药应用中大量先导化合物如抗生素、抗真菌剂和抗肿瘤剂等的重要来源。
尽管BGC潜力巨大,但实验验证证实只有一小部分(约3%)细菌基因组具有合成次级代谢产物的潜力。许多含有BGC的微生物由于非适应性表达、缺乏有利的诱导条件以及错综复杂的调控机制等原因,仍未被培养或不适合进行微生物遗传操作。异源表达是克服这些挑战的关键策略,找到理想的宿主往往需要复杂而特殊的条件,评估目标BGC与宿主的兼容性至关重要,因此研究BGC的水平转移具有重要的研究意义。
水平基因转移(HGT)是一种常见现象,基因组比较研究也发现HGT是BGC进化和适应的重要驱动力。目前已开发出多种计算工具来帮助检测HGT:GIST和IslandViewer利用基因组序列组成特征;DarkHorse和HGTector依赖序列相似性(最佳匹配);Ranger-DTL和AnGST采用系统发生学方法比对基因树与各自的物种树。
尽管在HGT检测方面取得了进展,但BGC的水平转移仍然是一个新兴的领域。在宏基因组组装基因组(MAGs)中,BGC的进化尚未完全定性。了解BGC的水平转移对于理解它们在自然环境中的传播和进化、确定具有特定生物合成能力的菌株之间的基因交换程度至关重要,并有助于选择最佳菌株用于异源表达。
本研究开发了水平转移生物合成基因簇搜索器(HTBGC-Finder),用于从MAGs中识别最近发生水平转移的生物合成基因簇(HTBGC)。HTBGC-Finder可以自动从MAGs中检测出潜在的HTBGC,安装和操作都很简便。利用这一工具,系统分析了人类肠道中潜在的BGC水平转移事件,深入了解了它们在个体内部的分布和进化情况。本研究加深了对微生物群落适应、资源利用和BGC转移的理解,为开发新的生物技术应用提供了重要的见解。
结 果
HTBGC-Finder的开发
HTBGC-Finder是一个计算工具,旨在从MAGs中识别个体中最近发生的水平转移BGC。图1是该工具的示意图,主要组成部分包括(1)BGC鉴定和分类特征识别:根据分类学特征构建非冗余MAGs,识别其中的BGC并确保分析的准确性。(2)构建基因簇家族(GCF)网络:根据BGC相似性聚类产生不同的GCF,便于研究BGC在不同分类群中的分布和进化。(3)识别离群BGC:离群BGC与同一GCF中的其他BGC在分类学上存在差异,被标记为潜在的横向转移BGC。(4)从参考基因组中识别BGC:追溯离群的BGC所在的MAG,并从美国国家生物技术信息中心(NCBI)检索与其分类学特征相匹配物种的参考基因组,鉴定这些参考基因组中的BGC。(5)根据MAG和参考基因组构建GCF:将GCF中识别出的离群BGC所在的MAGs及与离群BGC分类学特征相同的参考基因组进行分组分析,推断潜在的HTBGC。(6)系统发育分析:通过最大似然法(ML)进行系统发育重建,比较BGC与MAGs和参考基因组之间的系统发育距离,以进一步推断水平转移事件。
HTBGC-Finder将MAGs作为输入,以表格形式输出BGC的宏基因组来源、系统发育距离、统计p值、HTBGC可能性、BGC长度、类型、HTBGC的受体、潜在供体以及同属参考基因组内的相似BGC的信息(安装及使用见https://github.com/Shirly-Yang/HTBGC-Finder)。

图1. HTBGC-Finder流程概述
(A) HTBGC-Finder的输入文件包括宏基因组组装的基因组(MAGs)。(B) 下一步是根据平均核苷酸同一性(ANI)去除多余的MAGs。(C) 然后识别MAGs中包含的生物合成基因簇(BGC)。(D) 分析不同MAGs的分类学特征。(E) 下一步是根据BGC相似性构建基因簇家族(GCF)网络,并根据分类学分析确定每个GCF中的离群BGC。(F) 下一步是检索离群BGC对应的参考基因组。(G) 然后,确定参考基因组中包含的BGC。(H) 将离群BGC和从参考基因组中识别出的BGC聚类到GCF中,并将仍然离群的BGC视为潜在的水平转移候选者。(I) 对候选BGC中每个基因的系统发生关系进行统计分析。(J) 与MAGs的系统发育距离比参考基因组更近的离群BGC被推断为水平转移。
HTBGC-Finder的验证
为了评估HTBGC-Finder的识别性能,本研究应用该工具检测了人类微生物组项目(HMP)宏基因组样本中此前报道过的、可能参与硫肽生产的HTBGC。本研究从该文章中获取了与硫肽相关的原始测序数据,组装并分箱生成了1150个bins。随后使用HTBGC-Finder对数据集采用默认参数进行了分析,结果识别到一些潜在的HTBGC,其中包括来自SRS048870_bin18的硫肽(图2,表S2)。在1150个bins中,根据相似性将1840个BGC聚类为1035个GCF。值得注意的是,SRS048870_bin18中的硫肽BGC与SRS022071_bin51、SRS011084_bin14和SRS065504_bin21中的BGC划分为同一个GCF(图2a),具有不同的分类学特征:SRS048870_bin18为Bacteroidota,其他则为Bacillota。SRS048870_bin18的独特系统发育距离表明其BGC有可能是水平转移而来。
HTBGC-Finder进一步分析SRS048870_bin18与NCBI上与其分类学特征相匹配的23个参考基因组(表S3),识别所有的BGC并重构GCF,结果显示该硫肽BGC仍然是孤立的,没有与其他BGC聚类到任意一个GCF中(图2B),这进一步该BGC表明可能存在水平转移事件。
为了进一步研究这种可能性,HTBGC-Finder采用了系统发育分析,结果显示,与参考基因组相比,该硫肽BGC与MAGs的系统发育关系更为密切(图2C),且差异具有统计学意义(p < 0.001)(图2D)。这一结果表明,硫肽BGC是由水平转移而非垂直遗传而来的可能性更大。
此外,本研究还对参考基因组和与硫肽BGC聚类在同一GCF中的MAGs进行了平均核苷酸同一性(ANI)分析(表S4)。结果显示,硫肽BGC所属的SRS048870_bin18与Phocaeicola dorei DSM 17855T(RefSeq汇编登录号:GCF_000156075.1)的ANI最高(93.019%),这意味着Phocaeicola dorei 是与SRS048870_bin18最接近的物种(图S1)。
采用同样的方法,HTBGC-Finder从数据集中共鉴定出15个潜在的HTBGC,包括13种RiPP、1种PKS和1种归入“其他类”的多肽。对于RiPP,除了已鉴定的硫肽外,还鉴定出8个环内酯自诱导器(CLA)、2个含RiPP识别元件(RRE)、1个既含有CLA和又含有RRE的杂交肽和1个RiPP类似物(图2E,表S2)。在分类学上,10个BGC属于类杆菌(Bacteroidota,亦称Bacteroidetes),5个属于芽胞杆菌(Bacillota,亦称Firmicutes)(图2F,表S2)。
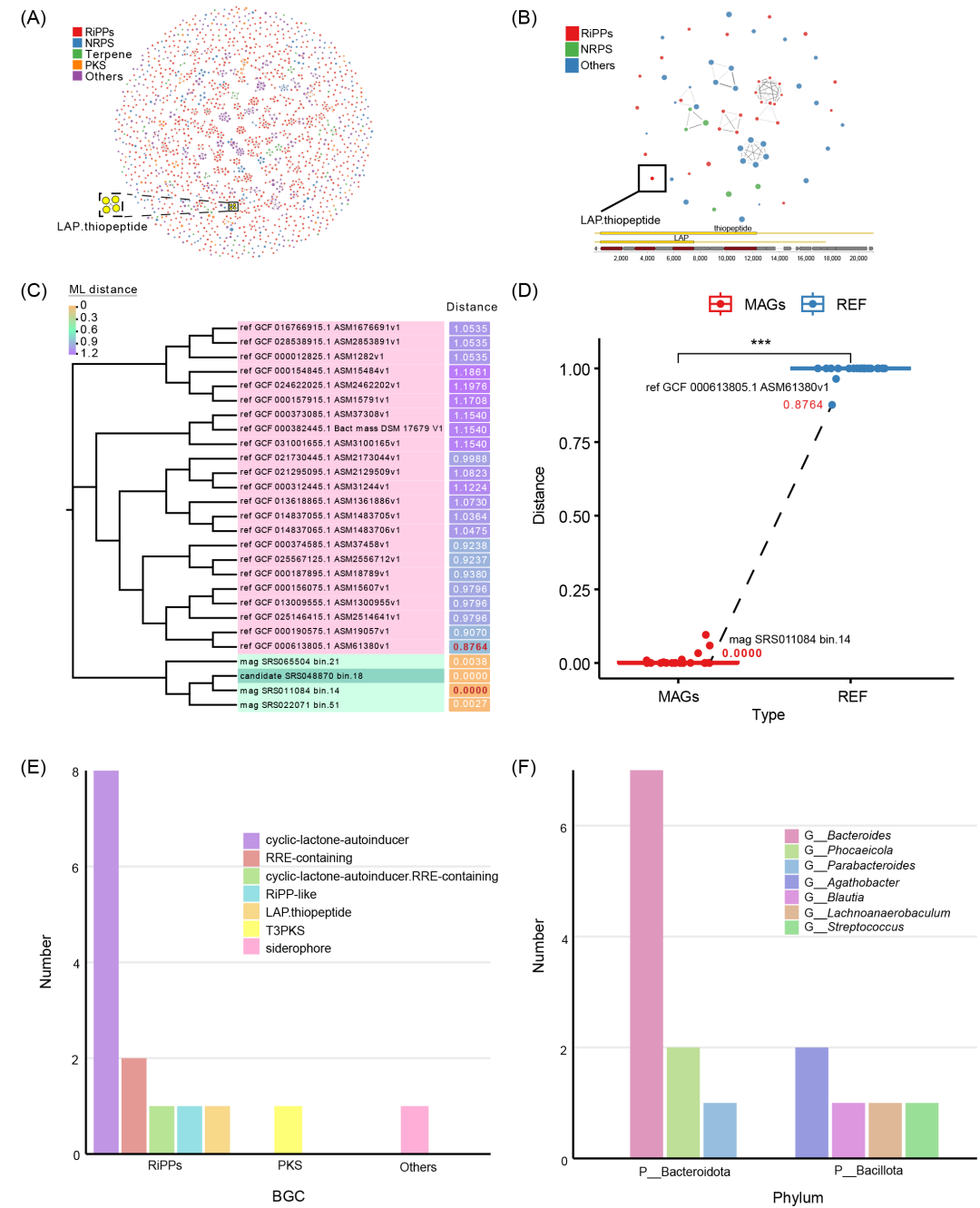
图2. 潜在横向转移的硫肽生物合成基因簇(BGC)的检测
(A)由从1150个宏基因组(MAG)中鉴定出的BGC构建的基因簇家族(GCF)网络。包含目标硫肽BGC的GCF用黄色标出。(B) 将目标硫肽BGC和BGC聚类在相应分类的参考基因组中得到的GCF网络。目标硫肽BGC包含12个基因。(C) 目标硫肽BGC的最大似然法(ML)系统发育分析。以SRS048870_bin18_contig135中硫肽BGC的第12个基因为查询基因,以同一GCF中相应分类群和MAG的参考基因组为查询群,进行ML系统发育重建。参考基因组用RefSeq基因组登录号和相应的组装号表示。宏基因组数据用MAG编号表示。(D) MAG与参考基因组的系统发育距离比较。每个点代表目标硫肽BGC的一个基因。“MAGs”代表来自 MAGs 的 BGCs,"REF "组代表来自参考基因组的 BGCs,“Distance”代表与目标硫肽BGC的系统发育距离。两组之间的统计差异通过配对Wilcoxon检验进行比较。*** p < 0.001.(E) 已确定的潜在水平转移BGC的类型。(F) 已确定的潜在水平转移BGC的分类剖析。RiPP:核糖体合成和翻译后修饰的肽;NRPS:非核糖体肽合成酶;PKS:聚酮合酶;RRE:RiPP前体识别元件。
HTBGC-Finder性能评估
为了进一步评估HTBGC-Finder的性能,本研究模拟了不同分类水平的BGC转移,并观察该工具是否能检测到预期的转移事件。具体来说,选择了人类肠道微生物组中关键物种的20个参考基因组,并模拟生成了发生BGC水平转移的宏基因组数据。使用HTBGCFinder分析该数据,将预测结果中的HTBGC与模拟生成的HTBGC进行比较,以评估HTBGC-Finder的预测性能。接收器操作特征曲线(ROC)表明,HTBGC-Finder具有较高的灵敏度和特异性,曲线下面积(AUC)为0.885(95%置信区间 [CI]:0.810-0.960。这表明HTBGC-Finder在识别不同宏基因组数据的HTBGC时,能较为有效地区分真阳性和假阳性(图3A,表S5)。
接下来本研究分析了HTBGC-Finder的准确性是否受到宏基因组固有特征(尤其是文库大小)的影响。结果表明,HTBGC识别情况的F1分数不受5000万、1亿和2亿文库规模的影响(Kruskal-Wallis检验,p = 0.052)(图3B,表S6)。这表明HTBGC-Finder的性能在不同的文库规模下保持一致,在处理不同测序深度的宏基因组时较为稳健。
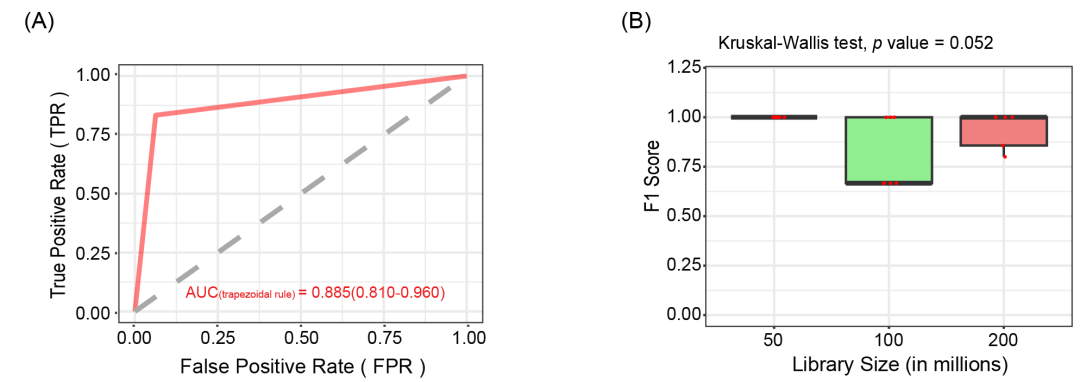
图3. HTBGC-Finder的预测性能
(A) 受试者工作特征曲线(ROC)显示了HTBGC-Finder对模拟宏基因组的预测性能,曲线下面积(AUC)(梯形法则)为0.885(95%置信区间[CI],0.810-0.960)。(B) 箱线图用于显示不同模拟宏基因组文库大小的F1分数分布。50M、100M和200M的文库大小的F1分数之间没有发现明显的差异(Kruskal-Wallis检验,p = 0.052)。
系统分析人类肠道微生物群中BGC的水平转移
为了深入研究人类肠道微生物群中BGC水平转移的复杂现象,本研究基于前人发表的人类肠道微生物群的大规模数据集(根据11,850个人类肠道微生物组数据重建了92,143个MAGs),利用HTBGC-Finder进行了分析。为了全面分析每个样本中BGC的水平转移情况,本研究根据生物样本登录号检索并分类,最终生成了5918个具有不同数量MAGs的合格样本(表S7)。在此基础上,选择了具有20个或更多MAGs的样本,系统地分析了其中潜在的HTBGC。
HTBGC-Finder共发现了81个可能在人类肠道微生物群中发生水平转移的BGC(表S8)。对这些BGC所属类群的分析表明,BGC水平转移仅发生在类杆菌(Bacteroidota,亦称Bacteroidetes)和芽胞杆菌(Bacillota,亦称Firmicutes),分别有50个和31个。从属水平上看,Bacteroides(81个中的19个)和Agathobacter(81个中的12个)的BGC水平转移发生率最高(图4,表S9)。
为了进一步探索潜在HTBGC的共同特征,本研究分析了它们的类型和功能。在已鉴定的81个BGC中,RiPPs是最主要的类型(81个中的76个),占比93.83%,其次是芳基聚烯(arylpolyene)(81个中的3个),占3.70%。此外,间苯二酚(resorcinol)和含有CLA和倍他内酯(betalactone)的混合BGC各占1.23%(图4)。从更具体的类型来看,含CLA和RRE的BGC占总数的近四分之三,分别为45个(55.56%)和14个(17.28%)(图4)。
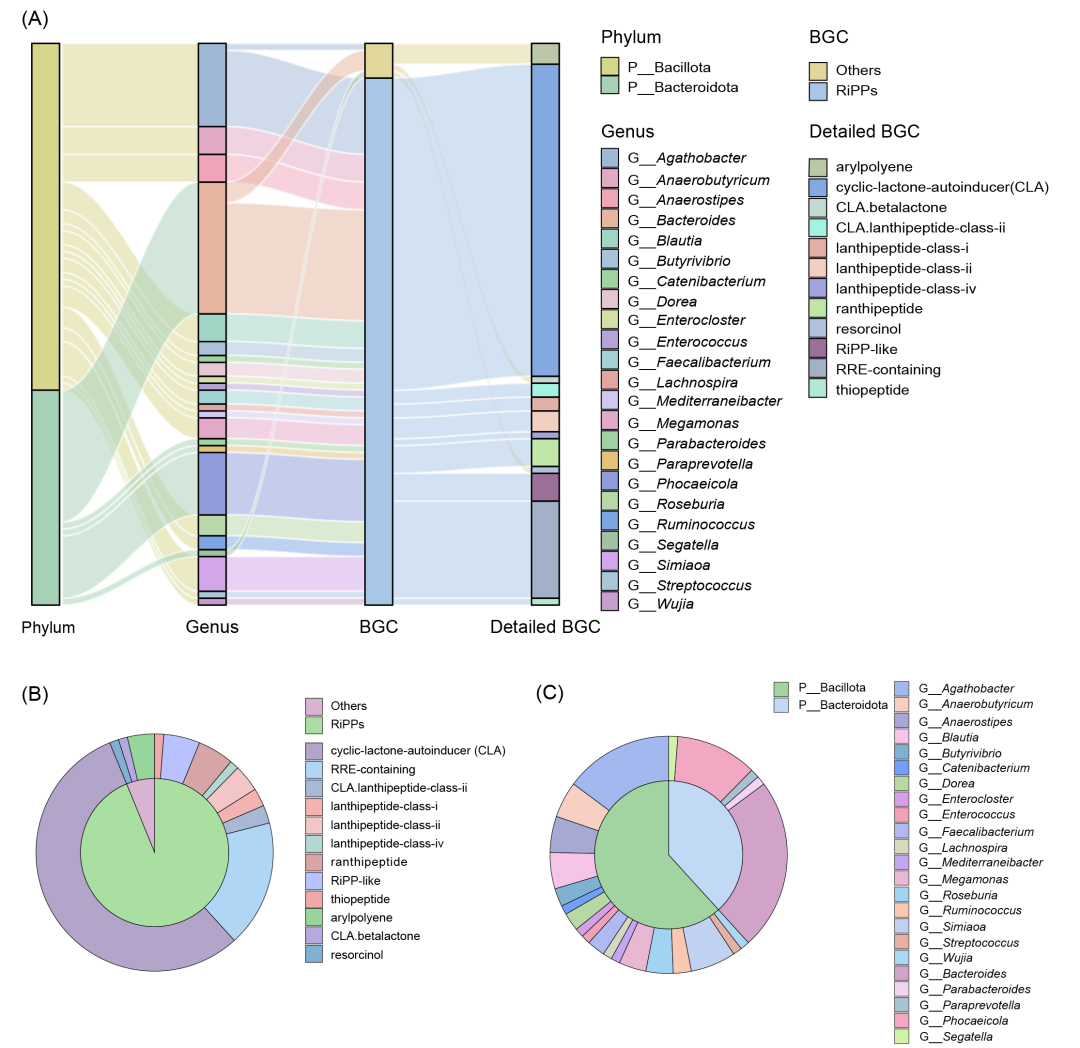
图4. HTBGC-Finder从人类肠道宏基因组中发现的潜在水平转移生物合成基因簇(HTBGC)
(A) 潜在HTBGC的组成及分类。(B) 潜在HTBGC的类型。(C) 潜在HTBGC的分类特征。RiPP:核糖体合成翻译后修饰的肽,RRE:RiPP前体识别元件。
为了全面了解BGC的水平转移现象,本研究对转移率进行了分析。结果表明,Bacteroidota(7732个中的30个)的水平转移率为0.39%,其次是Bacillota(22731个中的49个),为0.22%(图5A,表S9),不同门类之间的差异有统计学意义(Pearson's Chi-squared test,p < 0.001)。值得注意的是,Megamonas的转移率最高,为2.68%(112个中有3个),其次是Butyrivibrio,为2.22%(90个中有2个),Anaerobutyricum转移率为1.91%(209个中有4个),Catenibacterium为1.04%(96个中有1个),如图5B所示。Fisher’s exact test显示,各组之间的差异具有统计学意义(p < 0.001)。就BGC类型而言,RiPPs的水平转移率最高(27,152个中的77个),与非RiPPs组差异显著(Pearson's Chi-squared test,p < 0.001)(图5C,表S9)。此外,CLA和CLA相关(指含有CLA的BGC)的水平转移率最高(7267个中有48个,2.94%),与非CLA相关组相比,差异具有统计学意义(Pearson’s Chi-squared test,p < 0.001)(图5D,表S9)。
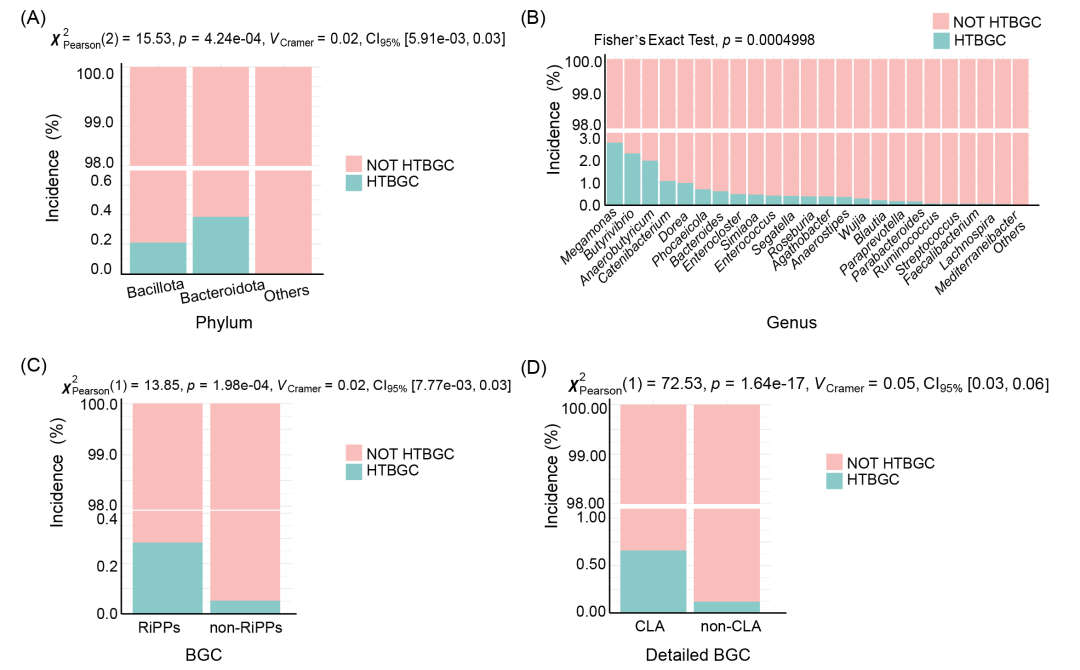
图5. 人类肠道微生物组中发现的潜在水平转移生物合成基因簇(HTBGCs)的发生率
(A) 不同门之间潜在HTBGC比较。(B) 不同属之间潜在HTBGC的比较。(C)核糖体合成和翻译后修饰肽(RiPP)与非RiPP的潜在HTBGC比较。(D)环内酯诱导器(CLA)与非CLA的潜在HTBGC的比较。NOT HTBGC组代表可能无法通过水平转移获得的BGC。HTBGC组代表被推断为水平转移而来的BGC。
为了进一步研究BGC水平转移的潜在模式并了解转移的趋势和方向,本研究还对水平转移BGC的受体和潜在供体进行了分析。结果发现,除了发生在同门水平之内的转移外,还观察到了Bacillota和Bacteroidota之间跨门水平的潜在水平转移现象,以及从双歧杆菌(Bifidobacterium)和脱硫弧菌(Desulfovibrio)向类杆菌(Bacteroidota)的转移(图6)。
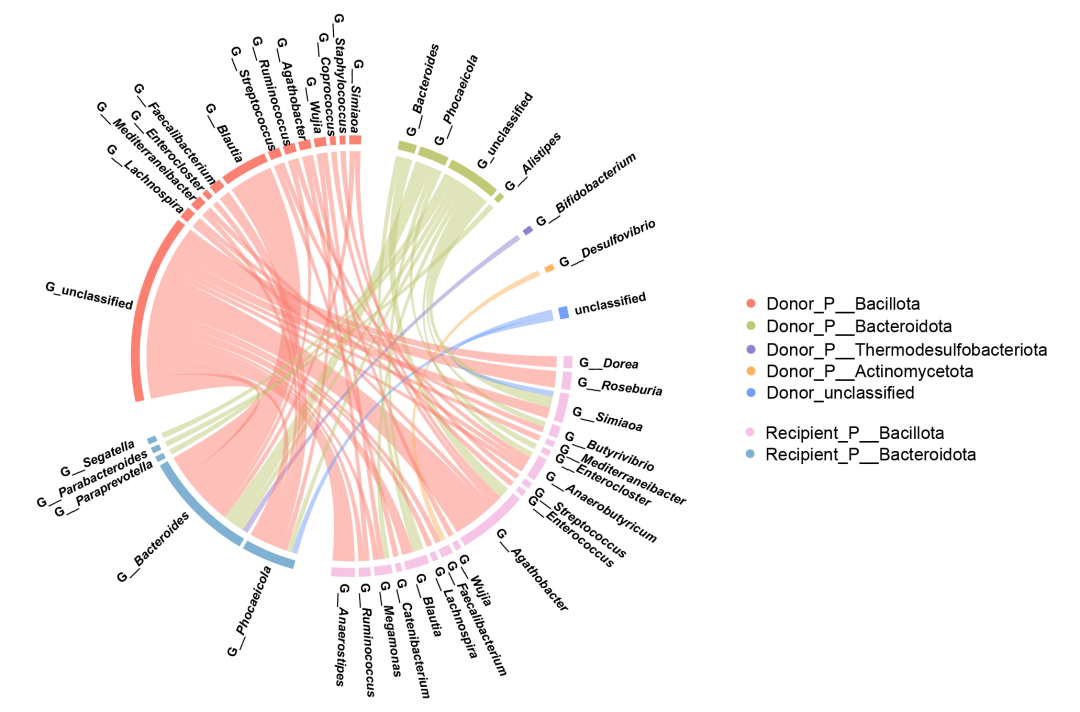
图6. 潜在水平转移生物合成基因簇(HTBGC)的受体和潜在供体
弦图显示了HTBGC的受体和潜在供体的分类特征。未分类的基因被归为“Others”。
讨 论
了解微生物群中BGC的自然水平转移对于阐明微生物群落动态、BGC进化及其生物技术潜力(如确定适合异源表达的宿主微生物)至关重要。宏基因组样本中密切相关的细菌之间的HGT可显著影响BGC的进化和传播,而生物信息学工具对研究这些过程至关重要。本研究介绍了HTBGC-Finder,旨在描述宏基因组数据集中的BGC水平转移事件。在评估HTBGC-Finder的性能时,生成reads和MAGs的模拟过程可能会带来固有偏差,包括组装过程中基因组恢复不完全、重新组装的MAGs中的BGC断裂以及读数模拟和分选造成的覆盖率不均。这些因素可能导致antiSMASH无法预测某些BGC,尤其是那些位于低丰度或高度片段化基因组区域的BGC,从而导致只有部分基因组的F1分数有效。因此,后续分析仅限于这些基因组。尽管如此,但对文库规模的比较显示,HTBGC-Finder的F1分数并没有受到测序深度的显著影响,这证明了它的稳健性。此外,ROC曲线的高AUC突出了HTBGC-Finder作为HTBGC检测可靠工具的潜力。
HTBGC-Finder识别HTBGC的能力为深入了解肠道微生物组提供了重要的信息。通过将HTBGC-Finder应用于人类肠道宏基因组,发现了81个潜在的HTBGC,证实了BGC水平转移的自然发生及其在扩大BGC多样性中的作用。研究结果表明,与其他BGC相比,RiPPs表现出独特的水平转移模式。RiPPs是模块化的大分子复合物,可促进酰基底物组装成多种生物活性肽,在微生物生物学和制药应用中发挥着至关重要的作用。有几个因素可能导致RiPPs在水平转移中的普遍存在:(1) 大小和复杂性:与其他BGC相比,RiPPs通常较小,复杂性也较低,因此更适合通过移动遗传元件进行水平转移。(2) 模块化结构:RiPPs的模块化性质允许交换特定的基因簇片段,从而提高了横向转移的潜力。(3) 遗传特征:RiPPs可能编码某些调控序列和移动元件等遗传元件,这有利于它们的水平移动。(4) 功能优势:RiPPs编码产生对细菌生存和环境适应至关重要的生物活性化合物,提供了促进其传播的竞争优势。
RiPPs的水平转移为合成生物学提供了丰富的研究材料。RiPPs转移与合成生物学的结合可在多个方面推动微生物疗法和药物发现,了解RiPPs转移机制还能为生态学和进化生物学研究提供新的视角,有助于揭示微生物群落如何通过RiPPs进行互动和适应,从而进一步研究它们在自然选择中的作用。此外,通过工程微生物释放特定的RiPPs,研究人员可以调节微生物群落的组成,促进有益微生物的生长,同时抑制病原微生物的增殖。这种基于RiPPs的疗法可用于治疗与微生物菌群失调有关的疾病,提供更精确的治疗方案。更重要的是,RiPPs的多样性使其成为新药开发的重要来源。通过结合合成生物学的方法,研究人员可以合成新的RiPPs或其衍生物,探索它们的生物活性,这不仅有助于发现新的抗菌剂,还可能开发出其他类别的治疗药物,如抗病毒、抗癌或免疫调节药物。
在各种类型的BGC中,CLA的潜在水平转移发生率最高。CLA与群体感应相关,这是细菌交换信息、协调行为和调节基因表达的一种机制,CLA编码的分子可作为自诱导物,向细菌的存活和密度调节发出信号。CLA的频繁水平转移表明,它们在促进微生物合作与协调方面发挥着至关重要的作用。通过调节群体行为和调控群体感应信号,CLA有助于提高微生物的适应性和生态平衡,从而增强微生物群落的恢复能力和功能多样性。
本研究表明,在人类肠道微生物群中占比近50%的芽孢杆菌(Bacillota)和类杆菌(Bacteroidota)中,水平转移BGC的发生率最高。这些菌群对肠道健康、碳水化合物发酵和短链脂肪酸生产至关重要。获得与群体感应相关的BGC可为这些细菌提供更强的通讯和代谢调节能力,有助于微生物群的稳定和宿主的健康。此外,这些具有高度水平转移倾向的细菌可能具有某些基因组特征,如重复序列和专门的基因转移系统,以促进这一过程。
虽然HTBGC-Finder通过比较基因组和系统发育分析在宏基因组数据中有效地识别了潜在的HTBGC,但必须承认它有几个局限性。组装的MAGs中可能包括原始微生物群中不存在的基因,从而可能导致BGC水平转移的错误结论;HTBGC-Finder依赖于注释完善的基因组参考库,这可能会限制其在分析注释不完善的细菌样本时的有效性;该工具并未考虑BGC的不完全转移或转移后重组等可能会影响功能完整性的情况。尽管存在这些局限性,但HTBGC-Finder在大规模鉴定潜在HTBGC方面取得了重大进展。不过,对水平转移过程的机理认识仍然很复杂,需要进一步的研究来完善方法并加深对微生物生态系统中BGC动态的了解。
结 论
本研究介绍的HTBGC-Finder可自动高通量识别MAGs中潜在的HTBGC,为宏基因组研究提供了一种用户友好且分析灵活的工具。该工具提供了检测水平转移BGC的可靠方法,使其成为研究微生物基因交换的重要辅助。本研究使用HTBGC-Finder对人类肠道微生物进行系统分析发现了81个潜在的水平转移生物合成基因簇,核糖体合成和翻译后修饰肽(RiPPs),尤其是与群体相关的环内酯自诱导器(CLA),表现出最高的转移频率。此外,芽孢杆菌(Bacillota)和类杆菌(Bacteroidota)为水平转移事件(包括跨门转移)最频繁的门类。这些发现揭示了微生物群落内部的遗传交流动态,有助于加深对肠道微生物群及其对宿主健康影响的了解。此外,这些结果对合成生物学、生物合成策略优化和药物开发也有意义。未来的研究通过阐明水平转移机制及其对微生物生态系统功能的影响,可以进一步完善对这些过程的理解,提高生物技术应用水平。
方 法
数据获取
根据Mohamed S Donia等人发表的文章,检索了宏基因组测序数据并收集了相应的元数据,详细的BioSample编号见表S1;根据Alexandre Almeida等人的文章检索了人类肠道微生物群数据集。
非冗余MAGs的分析
使用metaWRAP(V1.2.1),从所有样本的合并测序reads中构建MAGs。输入的测序数据通过模块“read_qc”分组进行测序读数质量的初步评估。使用模块“metaspades”进行组装,并根据模块“metabat2”确定不同的分箱。之后使用fastANI(V1.33),以99%的阈值去除这些MAGs中的冗余。然后使用antiSMASH(V6.1.1)识别BGC。接着使用kraken2(V2.1.2)以默认参数对每个MAG进行分类鉴定。最后,为了分析非冗余MAG中的同源BGC,使用BiG-SCAPE(V1.1.5)(截断值为0.3)将BGC聚类到GCF中,以便进行后续分析。
在GCF内识别离群值
根据GCF网络,利用不同BGC基因组之间的遗传距离矩阵确定离群BGC。具体来说,根据BGC基因组共享的最小相同类群来量化遗传距离。
对于两个基因组𝐵𝐺𝐶𝑖和𝐵𝐺𝐶j,遗传距离的计算基于它们在不同分类水平上的匹配度,包括域(D)、门(P)、纲(C)、目(O)、科(F)、属(G)和种(S),表示为taxo={D、P、C、O、F、G、S}。从最具体的层次(种)到最不具体的层次(域),检查每个分类层次的基因组之间是否匹配。
让index(t)表示分类级别𝑡在序列中的位置(例如,物种的索引设置为6,因为它是最具体的级别)。如果基因组𝐵𝐺𝐶𝑖和𝐵𝐺𝐶j在分类级别𝑡上匹配,它们的遗传距离计算公式为:

其中classification𝑖(𝑡)代表基因组𝐵𝐺𝐶𝑖在𝑡级的分类分类。
对于每个基因组𝐵𝐺𝐶𝑖,其与所有其他基因组的平均遗传距离计算公式为:
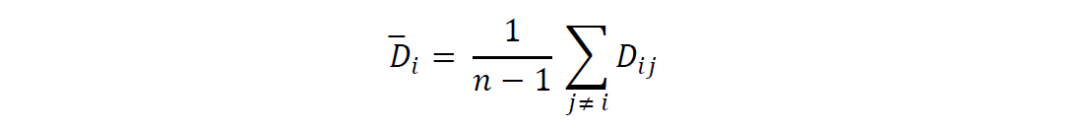
所有基因组的总体平均遗传距离计算公式为:
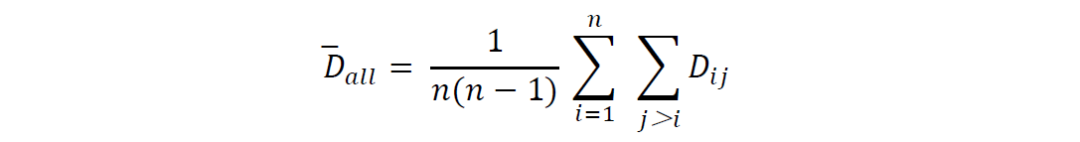
对于离群BGC的识别,基因组𝐵𝐺𝐶𝑖在以下情况下被视为离群:
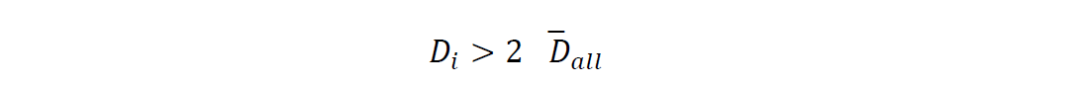
另外,也可根据Z值检测异常值:
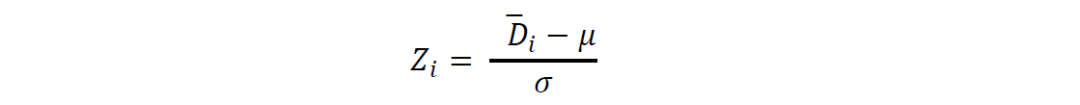
其中μ是所有平均遗传距离的平均值,σ是标准偏差,如果Z𝑖 > 2,则𝐵𝐺𝐶𝑖被视为离群值。
比较来自MAGs和参考基因组的BGC
使用ncbi-genome-download(V0.3.1)检索BGC源菌株属的参考基因组。这包括来自其种菌株的基因组和该属中所有经典菌株的基因组。由于不同分类单元的基因组数量不同,下载的离群菌株参考基因组的数量和类型也不尽相同。为了进行属一级的比较,下载了属内相同物种和经典菌株的基因组。当类型菌株较少时,研究人员可以通过修改命令行参数来调整下载的菌株。然后通过antiSMASH(V6.1.1)鉴定了参考基因组中的所有BGC。随后使用BiG-SCAPE(V0.1.5)比较了两者的相似性,参数与之前相同。
系统发育分析
目标BGC的进化关系是根据每个基因与相应参考基因组和分箱(bins)的DNA序列比对来评估的。具体来说,对于目标BGC的每个基因g,使用FastTree(V2.0.0)进行最大似然(ML)系统进化分析,以计算在参考基因组和bins中发现的同源BGC之间的系统进化距离矩阵:
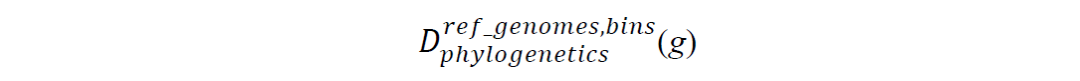
参考基因组和bins的距离矩阵中的最小值分别代表基因g的距离。对于无法在参考基因组或分类群中进行比对的基因,该基因与相应分类群之间的距离值被赋值为“1”,表示关系疏远:

同样,也得到了目标BGC中所有基因相对于参考基因组和分区的距离值集合:
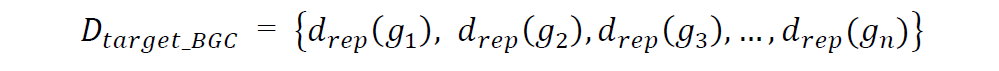
最后,通过配对t检验或Wilcoxon检验对两组(参考基因组和bins)之间的距离进行统计检验,以确定是否存在显著差异。
构建模拟HTBGC和宏基因组
模拟HTBGC和宏基因组的构建步骤如下。首先,从前人发表的文献中选出肠道中20个关键物种的参考基因组。其次,使用antiSMASH(V6.1.1)鉴定每个基因组的BGC组成。第三,每次模拟都选择10个参考基因组作为数据集,并随机选择10个从参考基因组中识别出的BGC,以2%的突变率插入到其他基因组中。不同大小的文库分别重复这一过程十次。第四,使用InSilicoSeq将插入后得到的基因组转换成双端宏基因组读数。InSilicoSeq根据默认的对数正态丰度分布和内置的其他分布生成多个基因组的读数,为每个基因组提供精确的丰度。通过不同的测序文库大小,共构建了30个模拟元基因组[(10个独特的集合)×(3种文库大小:5000万、1亿和2亿双端读数)]。
预测模拟宏基因组中的HTBGC
首先,利用metaWRAP(V1.2.1)默认参数从每个模拟宏基因组构建MAGs。然后使用HTBGC-Finder在生成的MAG中检测HTBGC。然后将检测结果与模拟插入结果进行比较,以评估HTBGC-Finder的预测性能。
用于评估HTBGC-Finder预测准确性的F1分数是根据HTBGC-Finder工具的组成部分antiSMASH成功识别的HTBGC计算得出的。F1分数被定义为精确度和召回率的调和平均值,计算公式如下:
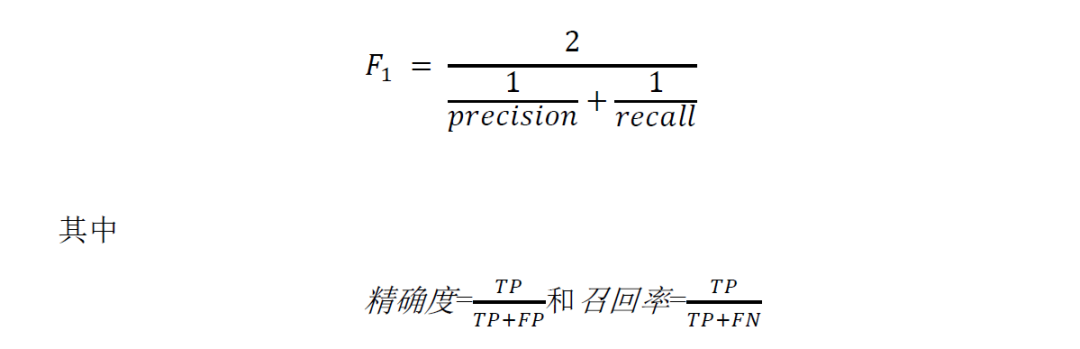
在这里,对于给定的宏基因组,TP(真阳性)是指被正确预测为水平转移的BGC的数量;FP(假阳性)是指被错误预测为水平转移的BGC的数量;FN(假阴性)是指被HTBGC-Finder遗漏的水平转移BGC的数量。
图表和统计分析
RStudio (V2023.12.1)运行R(V4.3.3)生成数据。正态分布变量的统计显著性采用学生t检验,非正态分布变量采用Wilcoxon秩和检验。根据具体分组条件,采用卡方检验和Fisher’s exact test分析。所有比较均为双侧比较,α水平为0.05。
GCF网络通过networkD3 (V0.4)、htmlwidgets (V1.6.4)和webshot (V0.5.5)可视化。进化树使用Evolview (V3)可视化。ROC曲线由pROC (V1.18.5) 绘制。方框图、柱状图和层饼图由ggplot2 (V3.4.2)绘制。冲积图由ggalluvial (V0.12.5) 绘制。通过ggstatsplot (V0.12.1)使用卡方检验或Fisher's exact test比较不同组间的潜在水平转移率,并通过ggplot2 (V3.4.2)和ggbreak (V0.1.2) 绘制图片。弦图用circlize (V0.4.16) 绘制。本研究使用的软件包均为免费开源软件。
代码和数据可用性
本研究主要数据和代码已上传至Github网站,网址为:https://github.com/Shirly-Yang/2025iMetaOmics。更详细的数据信息可联系通讯作者获取。补充材料(文本、图、表、中文翻译版本或视频)也可从线上(http://www.imeta.science/imetaomics/)获取。
引文格式:
Jiacheng Wu, Xiao Yang, Lanlan Zhao, Ziyun Li, Guoping Zhao, Lei Zhang. 2025. “Systematic Characterization of Horizontally Transferred BGCs in the Human Gut Microbiota Using HTBGC-Finder.” iMetaOmics 2: e62. https://doi.org/10.1002/imo2.62.
作者简介

吴嘉诚(第一作者)
● 山东大学公共卫生学院在读博士研究生。
● 研究方向为微生物的比较基因组学研究及方法开发,生物合成基因簇的挖掘,发表多篇SCI论文。

杨潇(第一作者)
● 山东大学公共卫生学院在读博士研究生。
● 研究方向为肠道宏基因组相关研究,生物合成基因簇的挖掘,发表多篇SCI论文。

张磊(通讯作者)
● 山东大学公共卫生学院教授,博士生导师。
● 研究方向为微生物组相关研究。泰山学者海外特聘专家,泰山产业领军人才,山东大学杰出中青年学者。德国不来梅大学微生物学博士(2005),美国UCLA博士后(2008)。2008年被UCLA聘为Faculty(Research Assistant Professor);2010年起任Principal Investigator。海外留学期间专注于微生物组研究,作为骨干承担3项NIH项目,作为PI主持2项美国政府研究基金,创建了唾液样本库和菌株库,参与构建了唾液多组学信息库,牵头多中心研究揭示了唾液转录组和微生物组与疾病关联(Gut, 2012; Gastroenterology, 2010等25篇SCI);获美国专利3项;获美国TRDRP基金会New Investigator Award和CBCRP基金会IDEA Award。归国后,率先开展山东微生物组研究,建立跨区域人群队列和样本库;创建了国家组学数据百科全书中最早、最大、用于标准化的微生物组数据集(7000例,526GB);系统开展了微生物组与健康的“相关→因果→干预”研究;研发了多款用于微生态干预的益生菌和益生元产品。围绕微生物组前沿方向,承担国家基金3项和省部级项目13项;发表SCI论文68篇,包括iMeta, 2024; Cancer Letters, 2024; Annals of Rheumatic Diseases, 2022; Gut, 2021; Pharmacological Research, 2020; GPB, 2019; Microbiome, 2018等。

赵国屏(通讯作者)
● 分子微生物学家,中国科学院院士,发展中国家科学院院士,美国微生物科学院院士。现任复旦大学生命科学学院微生物学和免疫学系主任;国科大杭州高等研究院生命与健康科学学院首席教授;中国科学院合成生物学重点实验室专家委员会主任。
● 研究领域涉及微生物基因组学、系统与合成生物学以及生物信息学等方面。曾参与启动中国人类基因组计划及相关生命“组学”研究,克隆若干遗传病致病基因;主持若干重要微生物的基因组、功能基因组、比较和进化基因组研究,解析SARS冠状病毒分子进化机制。在细菌蛋白质乙酰化组和肠道微生物组等领域作出若干开创性工作。组建并领导中国科学院合成生物学重点实验室,在酵母染色体重构,代谢组与代谢流量组研究,天然化合物细胞工厂制造,基因编辑技术研发等方向上,实现重要突破。近年来,参与组建并领导中国科学院上海生命科学研究院(现营养与健康研究所)生物医学大数据中心,为建设国家生物医学大数据综合性服务平台做好生物医学大数据的基础性工作。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics被DOAJ收录! (2025/1/27)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作
● iMetaOmics | 中科院动物所金坚石组综述16S rRNA基因扩增子测序技术的“前世今生”
● iMetaOmics | 浙大张天真组完成二倍体棉种泛基因组构建
● iMetaOmics | 张勇/李福平-先进糖蛋白组学在男性生殖研究中的潜在应用
● iMetaOmics | 暨南大学潘永勤/杨华组-炎症蛋白联合检测利于诊断甲状腺乳头状癌和结节性甲状腺肿
● iMetaOmics | 张开春组利用多组学方法揭示甜樱桃加倍后果色变化的候选基因
● iMetaOmics | 杜娟/林婷婷-慢性泪囊炎患者眼部菌群类型和纵向菌群变化
● iMetaOmics | 陈汉清/陈俊综述有关肝细胞癌治疗的新兴纳米医学策略
● iMetaOmics | 基因组所刘永鑫/卢洪评述微生物在提高杂种优势中的作用
● iMetaOmics | 上科大刘雪松组开发基于通路的肿瘤细胞鉴别工具TCfinder
● iMetaOmics | 中山大学刘鹏/邹宇田-整合人工智能实现HER2阳性乳腺癌精准管理
● iMetaOmics | 安徽农大李晓玉组-丛枝菌根真菌对玉米内生菌群的影响
● iMetaOmics | 徐涛/黄蓉/苏国海-急性冠脉综合征纵向多组学队列建设
● iMetaOmics | 通过整合宏组学促进人类与环境健康发展
● iMetaOmics | 苏州大学林俊组-揭示活性微生物及益生元/益生菌与关节炎联系
● iMetaOmics | 中国药科大学徐文波开发叶绿体基因组数据分析软件
● iMetaOmics | 清华刘晓组和复旦王久存组揭示特定细菌在皮肤老化中的作用
●iMetaOmics | 中南大学夏晓波团队揭示青光眼和SLE发病机制新关联
●iMetaOmics | 庐山植物园刘芬组揭示了自噬在植物-根微生物互作机制中的调控作用
●iMetaOmics | 杨瑞馥/袁静综述微生物组与“同一健康”的联系
●iMetaOmics | 同济/上海交大-开发支持群体分组分析的宏基因组测序综合分析软件
●iMetaOmics | 陈绍鸣-关于靶向NF-κB的潜伏逆转剂及其在HIV潜伏期的表观遗传和突变影响的评论
● iMetaOmics | 甘肃农大刘自刚组-强抗寒甘蓝型冬油菜的基因组组装和基因组特征解析
● iMetaOmics | 南京农大朱伟云组-外周血清素在结肠稳态中的作用
●iMetaOmics | 魏来/贾慧珏/何明光-多组学助力揭示塑造转录组的基因型-微生物组相互作用
● iMetaOmics | 徐州医科大学朱作斌组-微生物对寿命的调节:机制和治疗策略
● iMetaOmics | 白立景/邢凯组-解析脊椎动物肠道微生物多样性的影响因素
● iMetaOmics | 刘永鑫/陈同-用于食物微生物组成和时间序列研究的微生物组数据库FoodMicroDB
● iMetaOmics | 重庆大学王贵学组-肠道微生物细胞外囊泡在神经退行性疾病中的新作用及其治疗策略
● iMetaOmics | 四川大学王红宁组-解析产气荚膜梭菌的基因组宿主适应性
● iMetaOmics | 北京协和医院杨启文组-ramR基因突变增强免疫激活和依拉环素耐药性
● iMetaOmics | 香港中文苏奇组-抗菌多肽开发中的见解: 一个多学科视角的观察
● iMetaOmics | 上科大刘雪松组开发CD4 TCR特异性预测工具Pep2TCR
● iMetaOmics | 江苏省农科院植物细菌团队-解析中国梨火疫菌特征及溯源分析
● iMetaOmics | 基因组所刘永鑫组-易扩增子(EasyAmplicon):用户友好的扩增子测序数据分析指南
● iMetaOmics | 东京科学大学奥村学组-Hyena架构蛋白质语言建模
● iMetaOmics | 兰大南志标/段廷玉组-丛枝菌根网络影响邻近植物对病原菌的响应
● iMetaOmics | 汝之蜜糖彼之砒霜:源自益生菌LGG的研究证据
● iMetaOmics | 中国农大汪杰组-解析微塑料胁迫下玉米的分子响应
● iMetaOmics | 陈嘉莉/唐少军-奶源动物双歧杆菌乳亚种的膳食健康
● iMetaOmics | 西北农林曹阳春组综述艰难梭菌感染病理机制及饮食模式对其影响策略
● iMetaOmics | 宁波大学叶央芳组-解析驱动蟹肠道菌群稳定性的关键物种
● iMetaOmics | 中国农科院毕研亮组-精确消化道微生物群调节策略促进宿主健康
● iMetaOmics | 深圳大学李猛组-沉积物中古菌的多样性和代谢潜能
● iMetaOmics | 华东理工叶邦策组-多组学分析分枝杆菌侵染宿主过程
● iMetaOmics | 中国农业大学曹志军组-MicroRNA-微生物群互作调控宿主健康
● iMetaOmics | 中国农科院王秋霞组-微生物在土壤养分转换和作物营养代谢中的作用
● iMetaOmics | 军事医学研究院高月团队-提供高原心肌损伤新的治疗靶点
● iMetaOmics | 解放军总医院第一医学中心卫勃组-微生物与疼痛之缘
● iMetaOmics | 兰州大学刘鹏飞组-全球RNA病毒AMGs功能多样性
● iMetaOmics | 浙大袁长征组-解析MIND膳食模式的代谢组学特征及其与认知健康的关联
● iMetaOmics | 温州医科大李校堃院士团队-解析百合地黄汤重塑睡眠剥夺所致的肠-脑轴失衡
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://
www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









