






点击蓝字 关注我们
使用single cell analyst对多组学单细胞数据进行综合分析

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接: https://onlinelibrary.wiley.com/doi/10.1002/imt2.70038
● DOI: https://doi.org/10.1002/imt2.70038
● 2025年4月28日,中国医科大学李薛鑫等在iMeta在线发表了题为“Comprehensive analysis of multi-omics single-cell data using the single cell analyst”的文章。
● 本研究推出" The Single Cell Analyst "——一个基于网络的用户友好型平台,旨在助力多组学综合分析。该平台支持多种数据类型,涵盖六大单细胞组学技术:单细胞RNA测序(scRNA-seq)、单细胞转座酶可及染色质测序(scATAC-seq)、单细胞免疫图谱分析(scImmune profiling)、单细胞拷贝数变异检测(scCNV)、质谱流式细胞术(CyTOF)、流式细胞术以及空间转录组学,使研究人员无需编程基础即可完成整合分析。
● 第一作者:潘璐、汤步富、张旋
● 通讯作者:李薛鑫(xuexin.li@ki.se)、王振宁(znwang@cmu.edu.cn)、盛韧(shengren@mail.neu.edu.cn)
● 合作作者:Paolo Parini、Roman Tremme、Joseph Loscalzo、Volker M. Lauschke、 Bradley A. Maron、Paola Paci8, Ingemar Ernberg、Nguan Soon Tan、Ákos Végvári、 Zehuan Liao、Sundararaman Rengarajan、Roman Zubarev、范雨煊、郑旭、简新月
● 主要单位:中国医科大学、卡罗林斯卡医学院、东北大学、复旦大学、昆明医科大学、德国蒂宾根大学、哈佛医学院、罗马第一大学、南洋理工大学
亮 点
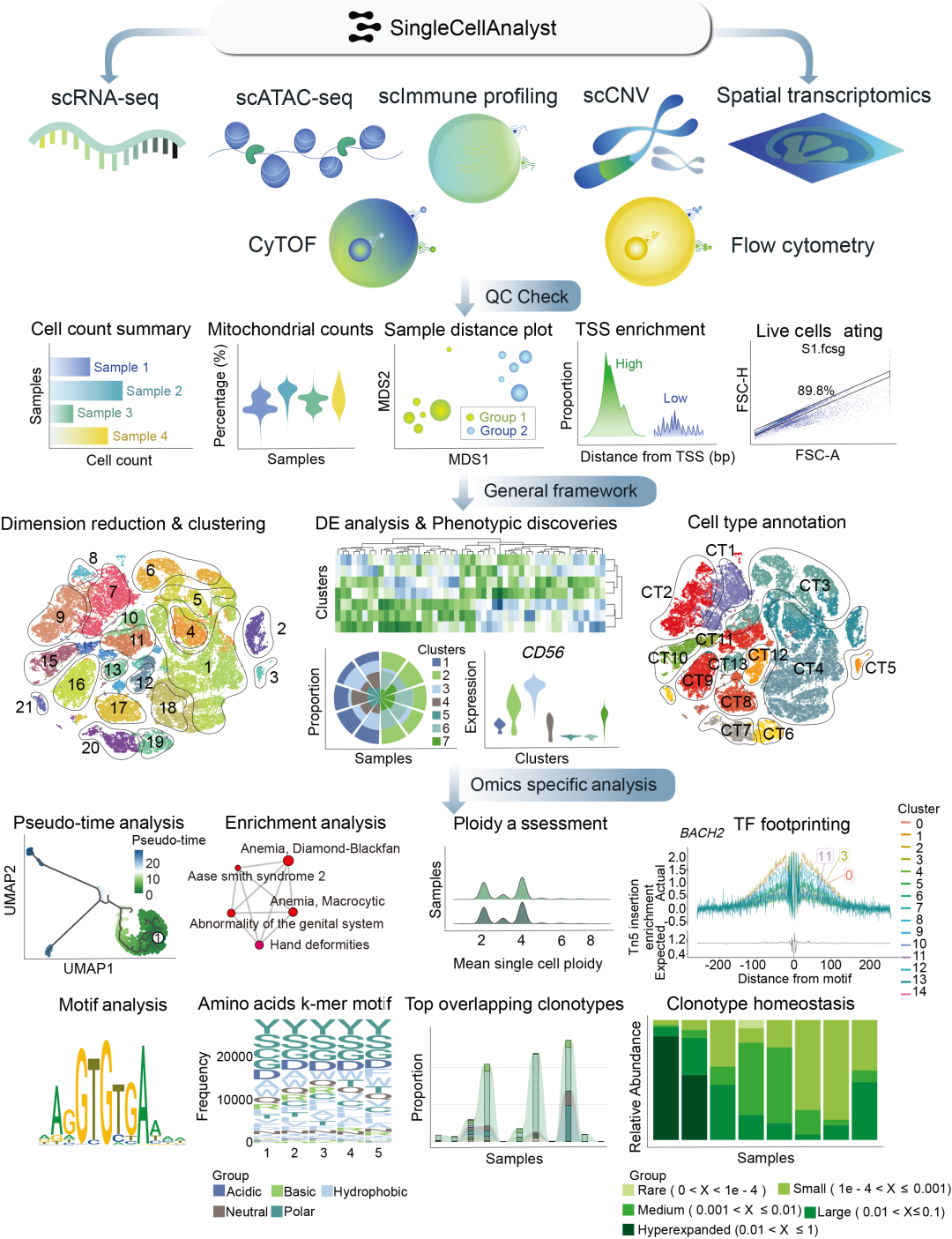
图1.single cell analyst平台服务器分析框架概述
● 全能分析平台:支持6大单细胞组学技术(scRNA-seq/scATAC-seq/scImmune/scCNV/CyTOF/流式细胞术)及空间转录组学分析;
● 零门槛操作:无需编程基础,直观友好的交互界面满足各层级研究者需求;
● 智能分析流水线:自动化完成质控、数据预处理、表型分析等关键步骤,一键生成可直接投稿的交互式图表;
● 极简工作流程:无需复杂生信背景,即可轻松完成高阶多组学整合分析。
摘 要
单细胞多组学技术的快速发展极大地增强了我们在前所未有的分辨率下研究复杂生物系统的能力。然而,许多现有的分析工具过于复杂,需要大量编程专业知识,这对计算能力不足的研究人员构成了障碍。为解决这一挑战,我们推出“The Single Cell Analyst”,这是一个用户友好的基于网络平台,旨在促进全面的多组学分析。“The Single Cell Analyst”支持多种数据类型,包括六种单细胞组学技术:单细胞RNA测序(scRNA-seq)、单细胞转座酶可及染色质测序(scATAC-seq)、单细胞免疫图谱分析(scImmune profiling)、单细胞拷贝数变异检测(scCNV)、质谱流式细胞技术(CyTOF)、流式细胞术以及空间转录组学,使研究人员无需编程技能即可进行整合分析。该平台提供在线和离线两种模式,为不同使用场景提供灵活性。它自动化了关键分析步骤,包括质量控制、数据处理和表型特异性分析,同时提供可交互的、可直接用于发表的图表。single cell analyst拥有20多种交互式中间分析工具,简化了工作流程,并显著降低了通常与类似平台相关的学习曲线。这一强大工具可适应不同规模的数据集,根据数据量在几分钟到几小时内完成分析,并确保计算资源的高效利用。通过简化复杂的多组学分析流程,single cell analyst为不同技术背景的研究人员提供了一个易于使用的全方位解决方案。
该平台可免费访问:www.singlecellanalyst.org
视频解读
Bilibili:https://www.bilibili.com/video/BV1HoGtzwEuR/
Youtube:https://youtu.be/bzIG3Zk58p8
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
单细胞组学技术的出现增强了我们在不同分子层面以单细胞分辨率解析生物现象的能力,并推动近年来单细胞多组学研究呈现指数级增长。这一趋势伴随着新型分析方法与工具体系的持续演进,以解析这些创新数据形式。然而现有平台往往存在应用范围与易用性局限——主要服务于单细胞(sc)RNA测序(scRNA-seq)等单一模态,且需要较高的编程技能。就基于网页的工具而言,目前尚缺乏能够跨组学,尤其是在当前主流的10X Genomics组学类型之间实现全套分析流程的综合性免编程解决方案。现有工具最多仅覆盖三种单细胞组学类型,部分工具对新用户而言存在较高使用门槛。
基于上述局限性,我们开发了Single Cell Analyst(www.singlecellanalyst.org)——一个支持六种单细胞组学类型全流程分析的在线平台,包括:单细胞RNA测序(scRNA-seq)、单细胞转座酶可及染色质测序(scATAC-seq)、单细胞免疫谱分析(scImmune profiling)、单细胞拷贝数变异(scCNV)、质谱流式技术(CyTOF)、流式细胞术以及空间转录组学。除广谱组学覆盖能力外,该平台还整合了20余种分步分析工具与可视化模块,进一步提升了分析灵活性。本技术方案基于我们前期发表的单细胞图谱和人类转录组细胞图谱(HTCA)工作构建,后者涵盖人类成体和胎儿逾百种组织类型。
该平台融合先进算法、交互式可视化与直观界面,使研究人员无需编程技能即可完成综合分析。下文我们将系统阐述该平台的核心功能、技术框架、基准测试结果以及证明其实用性的案例研究。
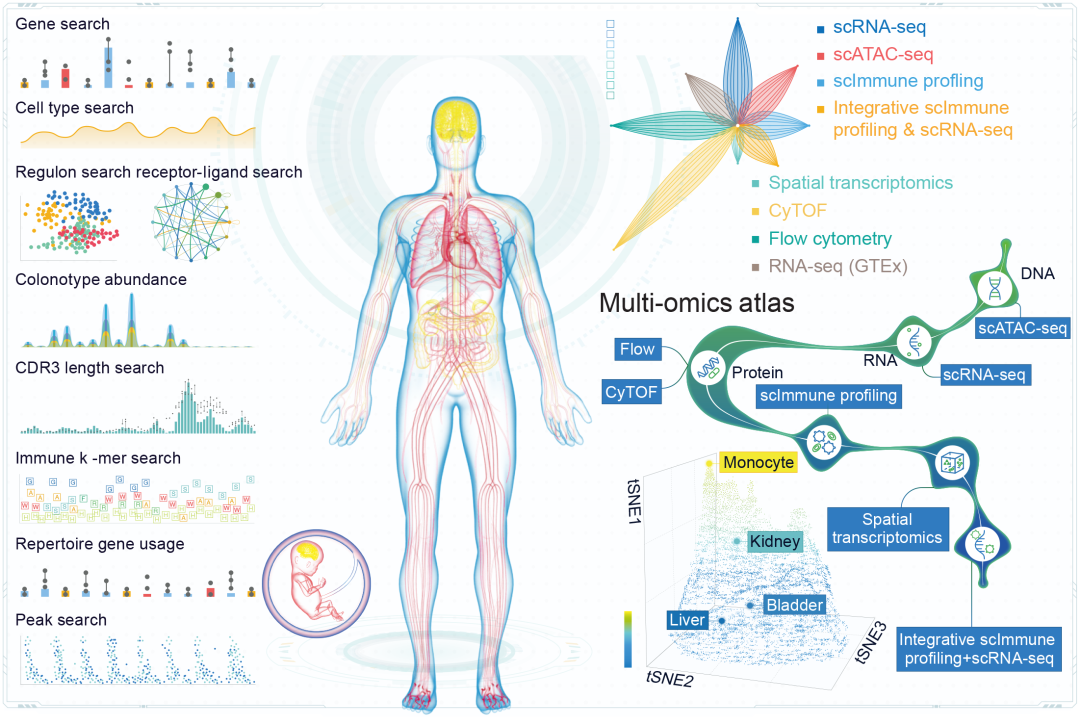
图2.单细胞分析平台数据库概览
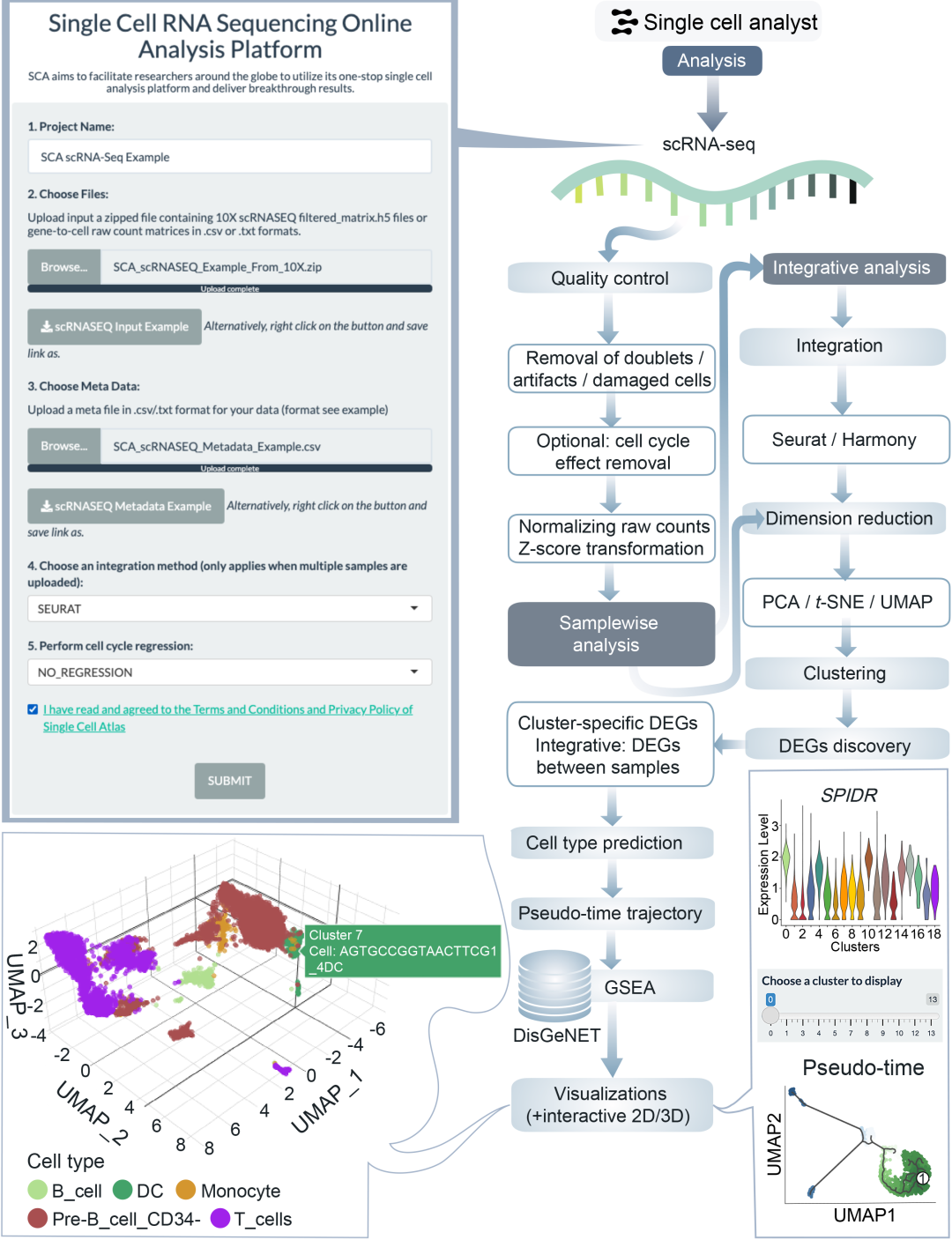
图3. ScRNA-seq分析流程框架图
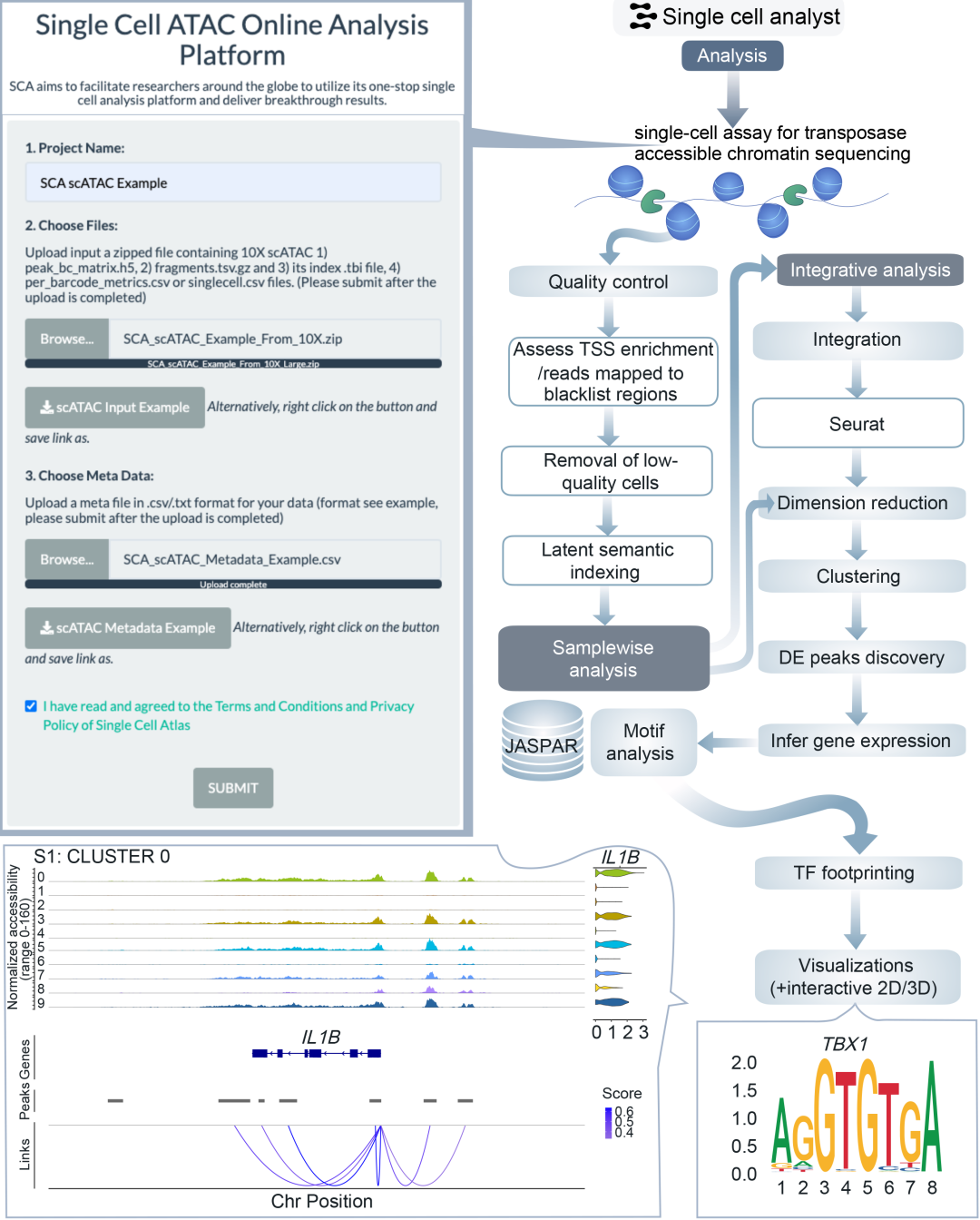
图4. ScATAC-seq分析流程框架图
结 果
Single cell ananlyst的特点
在scRNA-seq分析流程中,除质量控制(QC)与数据处理步骤外,还将进行细胞类型预测、差异表达分析、通路与富集分析,以及细胞簇和细胞类型的拟时序轨迹分析(图3)。用户可灵活指定任一细胞簇或细胞类型作为轨迹起始点。针对多样本数据,平台将启动整合分析以实现全面表型发现(图3)。
对于scATAC-seq数据,质量控制环节包含转录起始位点(TSS)评估。系统将展示各细胞簇的显著峰图及推断的基因活性(图1),同时进行motif足迹分析、富集motif鉴定,并通过计算顺式共可及网络(cis-co-accessible networks)识别共可及调控位点。多样本数据将执行合并分析(图4)。
在scImmune Profiling分析中,质量控制(QC)涵盖克隆型丰度与互补决定区3(CDR3)序列长度分布评估。分析流程还包括样本间受体库重叠检测、谱型分型、VDJC基因使用频率计算、克隆多样性评估、高频受体库筛选、氨基酸k-mer序列motif分析及克隆型稳态分析(图1)。若提交10X 5’ scImmune多组学数据,平台将启动整合分析——通过将受体库映射至对应scRNA-seq数据,评估推断细胞类型的克隆扩增情况(图5)。
scCNV分析流程包含ploidy评估与各细胞簇显著拷贝数变异(CNV)事件检测(图1)。分析过程中交叉验证bed文件与细胞身份摘要文件,为CNV位点分配差异ploidy值并过滤噪声细胞。利用选定CNV事件进行细胞层次聚类,对非二倍体细胞中特定CNV事件开展深入分析。聚类采用主成分判别分析(DAPC),基于主成分分析(PCA)结果通过贝叶斯信息准则(BIC)确定最佳簇数,聚类后生成可视化结果。平台将展示前50位CNV事件,并通过层次聚类识别潜在CNV簇。多样本项目将评估簇占比并构建系统发育树(图6)。
空间转录组学采用通用分析框架,通过差异表达分析(DE分析)识别各斑点簇的差异基因,并将降维与聚类结果叠加至空间图像以增强解读。提交多样本数据将触发自动整合分析。若同步提供scRNA-seq数据,则执行多组学联合分析。
CyTOF分析支持直接上传原始.fcs文件。多样本输入将启动整合分析,包括组间各标志物表达比较、基于非冗余评分的特征筛选、主次两级降维。首先通过元聚类(meta clustering)将表型相似细胞归并为100个超簇(完成第一维度降维),随后基于该结果进行次级降维与聚类。
对于流式细胞术数据,系统将从提供的原始.fcs文件中自动圈选单细胞事件,并剔除碎片或双联体细胞。进一步的质控包括样本距离分析,以评估组间和组内变异度并检测潜在离群值。此外,还将通过非冗余性评分对标记物的重要性进行评估。
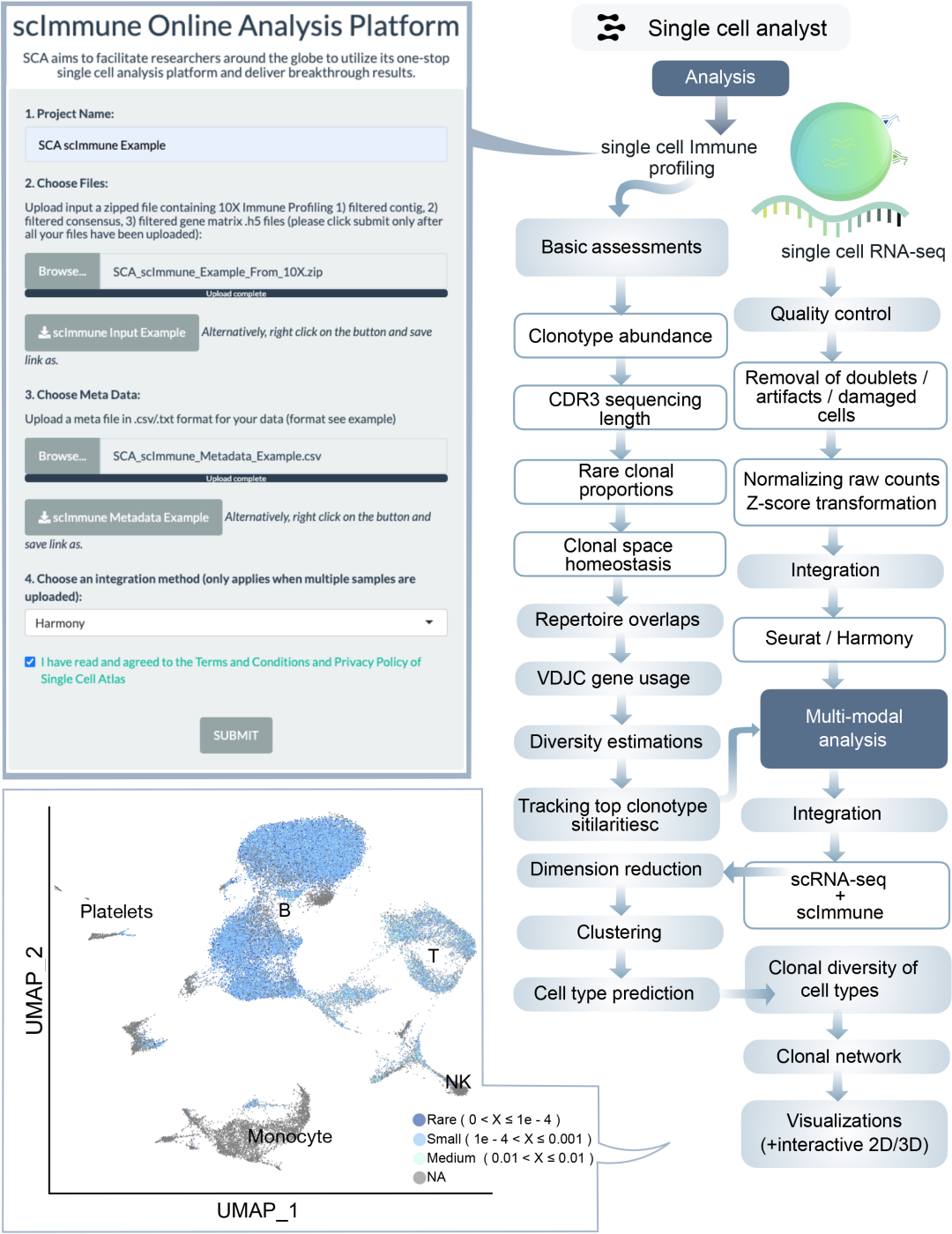
图5.单细胞免疫谱分析流程框架图
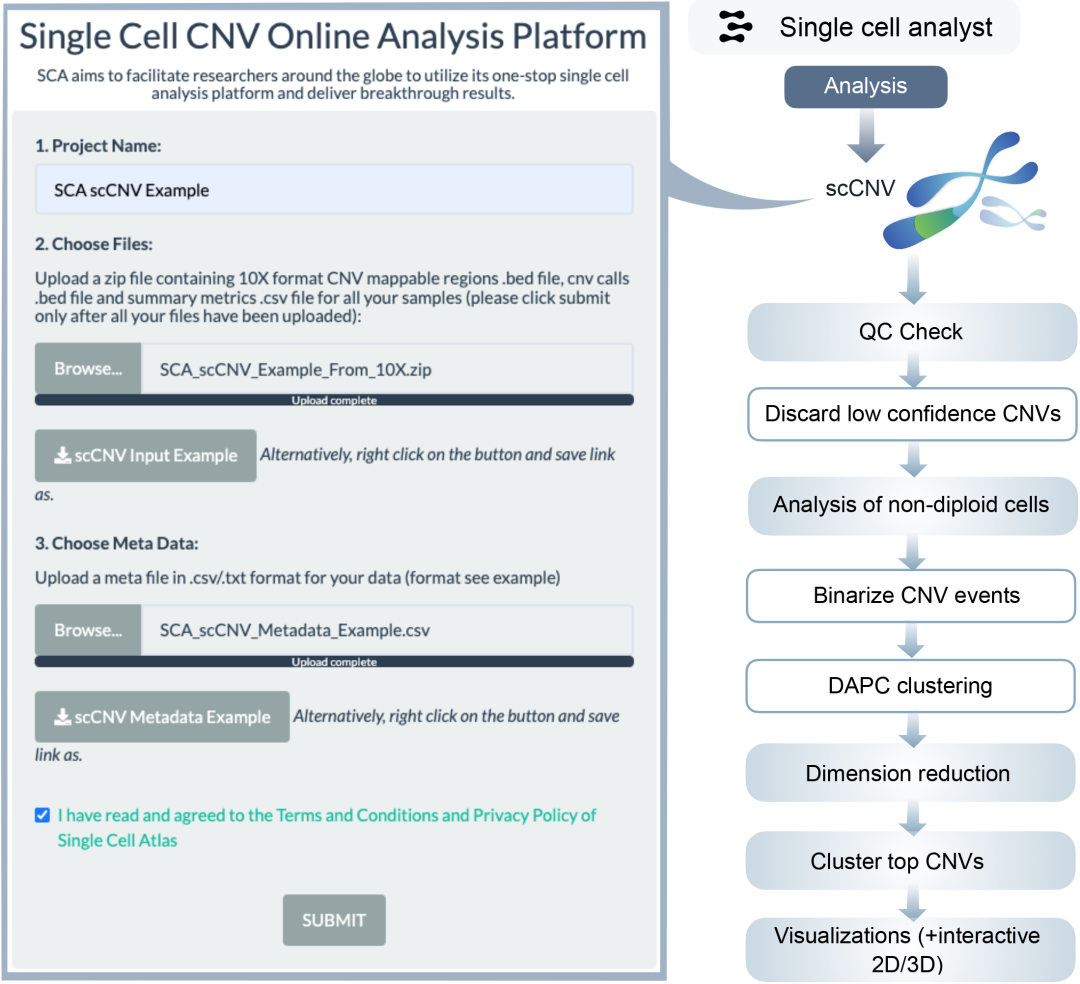
图6.ScCNV分析流程框架图
解决生物学问题的应用价值
Single Cell Analyst支持针对不同组学类型的多样化下游分析,以解答各类生物学问题。例如:在scRNA-seq流程中,聚类结果可帮助用户识别特定细胞群,差异表达分析能揭示与特定细胞类型或实验条件相关的基因,富集分析则将这些基因与通路和疾病关联,从而解析功能机制。拟时序轨迹分析可用于研究细胞分化或动态过程(如免疫细胞活化)。通过发表级图表和交互式工具可视化这些结果,研究人员能直观解读数据,从而解答诸如揭示肿瘤微环境异质性或识别疾病进展生物标志物等关键问题。
scRNA-seq分析
除标准QC与数据处理步骤外,该平台支持评估细胞类型预测、执行差异表达分析、开展通路与富集分析,并推断细胞簇或类型的拟时序轨迹。用户可根据生物学问题自定义拟时序分析的起始点。针对多样本数据集,平台执行整合分析以比较健康与患病样本或其他实验条件的表型差异。该流程特别适用于研究不同生物学背景下的基因表达动态与细胞状态转换。
scATAC-seq分析
在scATAC-seq流程中,TSS富集度与核小体条带模式评估等QC步骤,与可及染色质区域识别、motif足迹分析等下游分析相结合。通过计算基因活性分数,研究人员能将染色质可及性与基因表达关联。用户可探索顺式共可及网络与富集motif,解析驱动细胞功能的调控元件。多样本整合分析能揭示不同条件间共有的和特异的染色质特征。
单细胞免疫谱分析
scImmune Profiling流程专为免疫受体库分析设计,提供克隆型丰度、CDR3序列长度分布及VDJC基因使用频率等深度解析。平台支持受体库重叠分析、谱型分型与克隆多样性评估。对于多组学数据,整合scRNA-seq与免疫谱数据可将克隆型扩增映射至预测细胞类型,助力研究癌症免疫治疗或自身免疫疾病中的克隆扩增等免疫应答机制。
scCNV分析
该流程在单细胞层面解析拷贝数变异(CNV),通过识别ploidy水平并基于CNV谱聚类细胞,可检测具有特定基因组异常的子群。多样本分析中的系统发育树与层次聚类能鉴定样本间共有的和独特的CNV模式,尤其适用于CNV在肿瘤进展与异质性中起关键作用的癌症研究。
CyTOF分析
该流程通过降维与聚类从原始.fcs文件中识别细胞表型。用户可探索各簇的标志物表达、执行特征筛选,并评估样本对各簇的贡献度。元聚类(meta-clustering)将相似表型归并为更大簇群,有助于鉴定稀有细胞类型或状态,特别适用于研究复杂疾病或治疗反应中的免疫细胞异质性。
流式细胞术分析
流程自动化实施门控策略以排除碎片与双联体,继而降维聚类。通过非冗余评分评估各簇标志物表达以筛选最具信息量的标志物,多样本整合分析可解析群体水平变异与组间差异。该流程是需精确表型分析的免疫监测与生物标志物发现的理想选择。
空间转录组学分析
用户可直接在组织图像上可视化基因表达模式,将空间定位与分子特征关联。降维与聚类结果叠加至空间图像,以精细解析组织结构。多样本整合支持不同实验条件下组织架构的比较研究。当结合scRNA-seq数据时,多组学整合分析可提供空间分辨基因表达的整体视图。
服务器独立工具与其他功能
独立功能工具包含针对通用或特定组学数据集的分析模块与绘图选项(图S2),其参数可调以增强实用性。分析工具涵盖独立QC、降维、聚类、自动化细胞类型注释等步骤;可视化工具支持单细胞组学输出数据或原始输入数据(如CyTOF/流式FCS文件通道可视化)。
平台论坛页面支持用户交流、建议与咨询,推动SCA持续优化更新。这种互动模式旨在促进科学界更广泛地参与单细胞组学研究的全领域发展。
与其他方法的比较
在评估Single Cell Analyst与其他主流网络服务器的性能时,我们发现现有平台在分析步骤的完整性、组学类型覆盖度以及独立分析定制性(即可跳过连续分析步骤直接进入目标分析环节)方面均无法与本平台匹敌。尽管许多网络服务器(如ASAP、SingleCAnalyzer、Asc-seurat、Alona、SCTK、NASQAR、ICARUS)同时提供在线和离线分析功能,但其分析范围仍相对局限,这些平台仅支持scRNA-seq技术,且在质量控制、数据处理和表型评估步骤上仅有细微差异。此外,BIOMEX和SCIAp等分析平台将其服务范围扩展至两种单细胞组学类型;而ezSingleCell和Cellar则进一步支持scRNA-seq、scATAC-seq和空间转录组学三类组学数据。然而,上述网络服务器均未能达到Single Cell Analyst的全面性——本平台仍是目前唯一能够提供覆盖六种单细胞组学和空间转录组学的多样化分析流程,并兼容这些组学类型不同技术输出数据的解决方案。
Single Cell Analyst与现有工具的另一个显著区别在于其丰富的独立分析工具集。这一特性提供了独特的分析步骤独立性优势,用户无需按顺序完成一系列前置分析即可直接进入目标步骤。本质上,这意味着分析可以从中间步骤开始,从而显著提升了工具的整体灵活性。此外,Single Cell Analyst还具有一项其他网络服务器均不具备的独特功能:其与单细胞图谱数据库(SCA)(www.singlecellatlas.org)的深度整合。该数据库包含五大单细胞组学、空间转录组学以及bulk RNA-seq数据模块(图2),涵盖来自90多种不同成体和胎儿组织的正常样本。这一综合性资源为比较分析奠定了坚实基础,使用户能够深入探索其数据与广泛组织类型之间的关联。
实施本方案所需的专业知识
所有分析均通过基于网页的平台完成,该平台配备用户友好界面(UI)及简明的分步导航功能。虽然无需编程技能,但使用者须具备解读组学数据结果的基础知识。用户需将数据上传至我们的网络门户进行分析。因此,样本数量较多或文件体积较大的项目可能会经历较长的上传时间,且稳定的网络连接对确保分析过程不中断至关重要。为缓解此问题,处理大文件的用户可使用分析平台的docker版本,该版本支持离线运行,无需依赖稳定网络连接。此类分析的耗时取决于用户本地运行环境。关于docker版本的具体信息,请参阅代码可用性章节。
执行与可扩展性
可扩展性与性能测试
我们使用应用程序内提供的示例数据集,在Single Cell Analyst中测试了七种组学分析流程(scRNA-seq、scATAC-seq、免疫谱分析、scCNV、CyTOF、流式细胞术和空间转录组学)。针对更大规模的数据集,强烈建议在服务器或高性能计算机上通过docker部署运行。
性能测试结果
我们评估了不同CPU和内存配置下的性能指标(包括运行时间)。下表展示了分析流程的具体测试结果与配置建议。
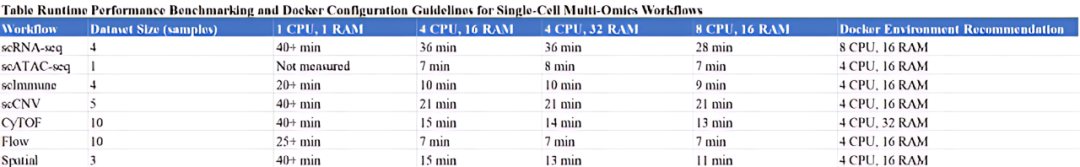
• 在线版本:最适合处理小于2GB的数据集,该版本运行于配备8GB内存且CPU数量可弹性调配的服务器。
• Docker版本:强烈建议用于超过2GB的数据集。对于更大规模的数据集,推荐在具有充足内存和CPU资源的系统上使用Docker版本。用户应根据表S3的推荐配置,在服务器或高性能本地计算机上运行该平台。
讨 论
该平台强大的功能与用户友好的设计,使Single Cell Analyst成为推动临床研究与诊断的重要工具。具体而言,该平台能够实现跨组学的疾病标志物识别、疾病异质性解析以及治疗耐药机制的探索。以癌症免疫治疗为例,Single Cell Analyst可鉴别区分治疗响应者与非响应者的分子特征,助力开发更有效的治疗方案。此外,其可视化工具能生成可直接用于发表的图表和临床级报告,促进研究人员与临床医生间的沟通。通过简化复杂的单细胞分析流程,本平台有助于推动精准医疗发展,并加速单细胞技术在临床诊断和治疗开发中的应用。
为保持平台的动态响应能力,我们在GitHub(https://github.com/singlecellanalyst/SCAWebserver)上进行版本控制,及时处理问题、优化功能并整合新特性。为满足用户需求,所有分析流程均已容器化(Docker),支持用户在本地或自有服务器上运行分析。各组学工作流程的详细说明与应用指南可从https://github.com/singlecellanalyst?tab=packages获取。我们致力于持续更新平台功能,确保Single Cell Analyst在这一快速发展的领域始终保持领先地位。我们认可现有诸多优秀工具各有优势,但坚信Single Cell Analyst的综合性能与持续优化的承诺,使其成为单细胞多组学分析领域独特而高效的选择。
针对可能的技术问题,平台提供多重支持机制:用户可通过"论坛"(https://www.singlecellanalyst.org/forum)或"联系我们"(https://www.singlecellanalyst.org/contact)页面进行问题讨论;GitHub仓库则用于问题追踪与解决。平台内置的错误处理机制会实时提示输入或格式错误,并指导修正措施,确保分析流程无缝进行。
当前版本主要面向人类数据分析,未来更新将扩展至其他物种。此外,平台升级将重点加强多模态单细胞数据(如同批细胞的scATAC-seq与scRNA-seq)的联合分析能力。由于此类分析需要较大计算资源,而在线版本受8GB内存限制,目前暂未实现该功能。虽然Docker版本技术上可行,但为保持初版一致性,我们计划在后续更新中为Docker版本增加联合分析功能。平台还将支持代谢组学等新型组学数据,以完善多组学分析体系。为增强数据整合能力,我们将开发更强大的多模态数据融合流程,并整合机器学习方法以提升预后与诊断分析功能。通过增加工作流定制选项、优化大规模数据处理能力,以及基于社区反馈的持续改进,Single Cell Analyst将始终为全球单细胞研究社群提供前沿、易用的分析解决方案。
结 论
为促进单细胞组学研究的可及性与高效性,我们开发了Single Cell Analyst——一个支持六种单细胞组学及空间转录组学的综合性多组学分析平台。该平台集成了主流单细胞组学分析流程,为缺乏深厚计算背景的研究人员提供了重要助力。通过免除依赖项下载需求并规避软件安装的复杂性,Single Cell Analyst有效降低了技术门槛,推动了单细胞技术的广泛普及。
本平台以高效性、用户友好性和广泛适用性著称,已成为单细胞分析领域的重要资源。其特色在于:流程化的工作流设计、关键步骤的自动化执行以及可一键生成发表级图表的功能,使实验科学家能够便捷开展多组学分析,惠及更广泛的研究群体。无需编程的操作模式与整合分析能力显著降低了技术壁垒,让不同背景的研究者都能轻松探索和解读复杂的单细胞数据。
平台跨组学数据集的分析能力促进了生物现象的系统级认知,为多组学研究提供了前所未有的灵活性。这种能力不仅加速了科学发现进程,更助力研究者深入解析细胞异质性、组织结构和分子机制。通过为生物医学复杂问题提供创新解决方案,Single Cell Analyst不仅推动了单细胞技术的应用普及,更拓展了其应用场景并促进跨学科合作,标志着单细胞研究领域的一项变革性进步。
方 法
分析框架
该平台支持对定量后的原始文件进行标准化流程分析,包含自动生成交互式可视化结果的完整分析流程。用户上传必要的输入数据和样本信息表后,通过单次点击即可触发下游分析(图3-6)。以下将详细介绍各特异性组学分析流程。
单细胞RNA测序(scRNA-seq)分析框架
本分析框架专为10X Genomics平台定量后的scRNA-seq数据设计,可直接使用其定量输出作为输入文件。同时,该框架也支持其他技术平台生成的基因-细胞表达矩阵(csv或txt格式)(图5)。该分析支持多个scRNA-seq样本的整合分析,并整合了seurat和monocle3等流行软件包的功能。
质量控制检查
对于每个提交的项目样本,在进行质控过滤前将计算一系列质控指标。平台将计算每个细胞中线粒体基因的表达百分比,用于评估样本中可能存在的损伤细胞或实验假象。同时,还将计算每个细胞中检测到的基因数和RNA分子数。为了观察样本中可能存在的细胞周期效应,平台将使用样本中的高变基因对数据进行标准化和缩放。随后基于以下两组特征分别进行PCA和UMAP分析:1)细胞周期标志物(S期和G2M期标志物);2)高变基因,以比较细胞周期标志物对数据的影响。如果在样本中观察到明显的细胞周期异质性,用户可根据具体研究问题选择是否进行细胞周期回归分析。平台提供两种回归选项:双相回归和相位差回归。前者将消除所有细胞周期阶段的影响,后者仅消除G2M期与S期之间的差异。过滤前的质控结果将以可视化形式呈现给用户。
过滤后质控、降维与聚类分析
随后对每个样本进行过滤处理,保留表达基因数在200(不含)至25,000(含)之间且线粒体基因表达比例低于5%(不含)的细胞。此步骤可去除实验过程中可能产生的空微滴、双联体、假象和损伤细胞。样本数据将基于过滤后的数据进行标准化,并使用高变基因进行缩放。根据用户是否选择进行细胞周期回归,平台将执行相应的回归分析以消除细胞周期效应。过滤后相同的质控指标将再次展示给用户。对于每个样本,将使用过滤数据中的高变基因计算PCA、UMAP和t-SNE投影。基于前30个PCA维度计算k近邻(k-NN)和共享最近邻(SNN),并使用seurat中的FindClusters函数以0.8的分辨率进行聚类分析。聚类结果将通过静态2D和交互式3D的UMAP及t-SNE图展示。
基于聚类结果的差异表达分析
平台将使用seurat中的Wilcoxon秩和检验对每个簇和基因进行差异表达分析。设定差异分析的阈值为:基因在任一比较细胞群中的检出比例不低于10%。最终保留的差异表达基因(DEGs)需满足Bonferroni校正后p值<0.05,且该基因在目标簇细胞与其他簇细胞间的平均log2倍数变化的绝对值>0.25。计算DEGs在样本对数标准化数据中的中位表达量,并以热图形式可视化。同时绘制top基因的密度分布图。降维和聚类后的结果将通过UMAP、t-SNE和PCA可视化展示。每个样本各簇的DEGs将以表格形式展示,便于用户用于簇的细胞类型注释。
为辅助细胞类型注释,平台将基于聚类结果使用SingleR进行自动化细胞类型注释,并以人类原代细胞图谱作为参考数据集。自动化注释通过将每个细胞的基因表达谱与参考数据集(包含人体各种细胞类型的表达谱)进行相关性比较,根据最高相似度分配最可能的细胞类型标签。需要注意的是,自动化注释结果仅供参考,建议用户基于DEGs进行手动注释验证。由于注释准确性高度依赖参考数据集,参考数据中未包含的稀有或组织特异性细胞类型可能导致注释错误。因此,强烈建议用户结合差异表达基因和生物学背景进行手动验证以确保结果的可靠性。为辅助手动验证,平台将计算各注释细胞类型中top DEGs的中位表达量,并以热图形式呈现给用户。最终的注释结果将通过静态2D和交互式3D的UMAP及t-SNE图展示。
富集分析
对于每个样本的各个细胞簇,平台将选取平均log2倍数变化值最高的500个差异表达基因(DEGs),基于DisGeNET数据库并使用DOSE软件包进行疾病-基因关联富集分析。保留校正后p值<0.05且Q值<0.05的富集疾病条目,针对样本中的每个细胞簇,分析结果将以柱状图、点图和热图等多种形式展示,满足不同的可视化需求。平台将构建基因-概念网络来展示基因与富集条目之间的关联关系,并通过网络图呈现每个细胞簇和样本的分析结果。同时展示疾病条目的富集网络,比较样本内各细胞簇间的疾病条目差异,并显示按细胞簇聚类的疾病条目网络。通过Upset图可视化DEGs与富集条目之间的关联关系。此外,对每个细胞簇的DEGs进行基因集富集分析(GSEA),结果以富集图形式呈现。最终,细胞类型标签将被映射到t-SNE和UMAP可视化结果中展示给用户。
拟时序轨迹分析
采用Monocle3进行拟时序分析。首先基于样本UMAP构建主图,为最大化结果灵活性,将每个细胞簇均作为拟时序计算的起始节点,所有计算结果都将返回给用户,便于用户根据具体生物学问题选择合适的拟时序轨迹。结果以交互式形式展示。此外,基于PCA、UMAP和t-SNE排序首维嵌入计算的拟时序结果也将结合聚类结果和注释细胞类型进行展示。
整合分析
对于提交的多样本项目(如健康与疾病样本比较),平台将执行样本整合分析。用户需在提交的元数据中提供分组信息。根据选择的整合方法(seurat或Harmony),采用不同整合策略:若选择seurat整合,将每个样本视为一个批次进行严格整合,适用于批次内变异较大的项目。通过筛选跨样本变异最大的特征作为整合特征,基于互惠PCA(Reciprocal PCA)进行降维,识别样本间互为近邻的细胞对(即互最近邻),剔除低置信度锚点后,根据样本对间细胞的共享近邻重叠度去除离群值,最终确定用于整合的锚点集。基于锚点进行样本对间迭代整合,对整合数据集进行缩放后,基于整合数据集的高变基因计算PCA嵌入,并通过UMAP和t-SNE进行降维。若选择Harmony方法,则根据用户提供的批次信息,基于预计算的未校正PCA嵌入进行整合:将细胞分配到各簇并计算每个样本在簇中的质心,计算各簇的校正因子使样本质心聚拢,根据簇特异性校正因子调整细胞在PCA嵌入中的位置,迭代至收敛。随着样本量增加,Harmony具有更快的时间复杂度。最终返回基于未校正PCA嵌入的Harmony校正嵌入,用于后续UMAP和t-SNE降维。整合前后的质控指标将通过UMAP、t-SNE和PCA可视化展示,作为整合质量的评估依据。
对整合数据采用k-NN和SNN模块化优化进行无监督聚类(seurat,分辨率0.8)。对整合数据集的每个细胞簇进行差异表达分析:与样本水平分析类似,使用Wilcoxon秩和检验,保留Bonferroni校正后p<0.05且绝对平均log2倍数变化>0.25的基因作为DEGs。计算DEGs在样本对数标准化数据中的中位表达量并以热图展示。基于聚类结果使用SingleR对整合数据集进行自动化细胞类型注释(参考人类原代细胞图谱),计算各注释细胞类型中top DEGs的中位表达量供验证,同时计算并展示各簇的样本组成比例。
整合数据集中的组间比较
对于提供多组别的样本,在每个细胞簇内进行组间比较,使用Wilcoxon秩和检验发现簇内组间差异表达基因(保留Bonferroni校正后p<0.05的DEGs),细胞数少于3组的比较将被跳过。展示DEGs在簇间和组间的表达分布。
整合数据集的拟时序轨迹分析
同样使用Monocle3,基于整合UMAP嵌入构建主图。将每个细胞簇或注释细胞类型作为拟时序起始点计算相应轨迹,用户可根据生物学问题选择合适的拟时序结果,交互式展示分析结果。
单细胞ATAC测序(scATAC-seq)分析框架
本分析框架专为10X Genomics平台定量后的scATAC-seq数据设计,可直接使用Cellranger的输出作为输入文件。由于scATAC-seq样本数据量较大,且受内存限制,目前暂不支持多模态整合分析。用户需在元数据文件中注明所使用的参考基因组版本。相较于其他组学分析,scATAC-seq数据的处理时间通常更长(图6)。
过滤前质量控制检查
使用Signac软件包计算并可视化每个细胞的核小体条带模式。计算转录起始位点(TSS)富集分数以评估TSS区域的富集水平,该分数与实验质量成正比。通过检测峰区片段数量评估细胞测序深度和复杂度,过低或过高的峰区片段数分别提示测序深度不足或存在双联体。另一项质控是识别黑名单区域(ENCODE定义的信号假象区域)读取比例过高的细胞,计算每个细胞中映射到黑名单区域的reads占总reads的比例。
过滤后质控、降维与聚类分析
基于质控指标,每个样本保留满足以下条件的细胞:1)峰区片段数>3000且<20,000;2)核小体信号<4;3)TSS富集分数>2;4)黑名单区域比例<0.05;5)峰区reads比例>15%。该过滤过程可去除信号异常的细胞以及测序深度不足或双联体等低质量细胞。随后使用TF-IDF方法进行标准化,该方法通过校正细胞测序深度,同时赋予稀有峰更高权重来校正峰信号。与scRNA-seq不同,scATAC-seq的特征选择将选取样本中前25%的峰。对标准化数据使用选定峰进行奇异值分解(SVD)降维,生成一组LSI(潜在语义索引)成分用于后续降维步骤。由于第一LSI成分通常反映技术变异而非生物变异,将予以剔除,使用第2至30个LSI成分进行UMAP和t-SNE降维。降维后基于第2-30个LSI成分进行k近邻(k-NN)和共享最近邻(SNN)分析,随后使用SLM算法进行模块化优化聚类。
基于聚类结果的差异可及性分析
使用Signac中的逻辑回归对每个簇和每个峰进行差异可及性分析,以峰数量作为潜变量减少测序深度影响。剔除在任一组细胞中检出比例<5%的峰,保留Bonferroni校正后p<0.05的差异峰作为最终结果,以表格形式展示给用户,同时提供UMAP和t-SNE形式的降维与聚类结果可视化。
从scATAC-seq推断基因活性及motif分析
为关联基因启动子可及性与基因表达,使用Signac统计每个基因上游2kb区域的片段数,作为该基因在各细胞中的活性分数。对基因活性矩阵进行标准化和缩放后,基于前述聚类结果,采用Wilcoxon秩和检验比较簇内细胞与簇外细胞的基因活性差异。剔除在任一组细胞中检出比例<50%的基因,保留Bonferroni校正后p<0.05且平均log2倍数变化>0.25的差异基因。计算每个峰的GC含量、可及性和长度,对差异分析得到的top基因与邻近峰进行相关性分析。每个簇中top基因及其上下游10kb区域内top峰的可及性信号将以覆盖度图形式展示。随后进行DNA序列motif分析:首先从JASPAR数据库查询motif位点并映射到峰区,对差异基因进行motif富集分析并可视化位置权重矩阵,绘制前6个差异基因对应motif的足迹图。
整合分析
对于多样本项目,将合并所有样本的峰区并进行类似单样本的特征选择,经TF-IDF标准化后通过SVD降维获取嵌入向量。使用第2-30个LSI成分进行UMAP和t-SNE非线性降维,基于相同成分进行k-NN和SNN分析后采用SLM算法聚类。对整合数据集进行差异分析获取top峰,并推断基因活性,绘制top峰与top差异基因的覆盖度图。
单细胞免疫谱分析(scImmune profiling)框架
本分析框架专为10X Genomics多模态定量后的scImmune profiling样本设计,支持同时提交多个多模态样本。若同一样本同时包含T细胞和B细胞受体库,需在用户提供的元数据中完整标注。需提交经过滤的数据文件(即Cellranger定量步骤的默认输出)。分析将基于CDR3核苷酸序列、氨基酸序列、VDJ基因和CDR3核苷酸序列进行克隆鉴定(图5)。
基础统计与克隆特征分析
针对每个样本的各个受体库,将计算并可视化基础统计量:包括独特克隆型数量、克隆丰度分布以及CDR3序列长度分布。通过分级统计方法评估受体库组成:首先按克隆型比例排序后分级(第一级包含前1-10克隆型,第二级11-100克隆型,以此类推),这种呈现方式通过对数化处理突出高丰度克隆型。其次分析稀有克隆比例:顶级分组包含单细胞稀有克隆型,次级包含2-3细胞的克隆型。同时通过五级分组评估克隆稳态:稀有组(0<X≤1e-4)、小组(1e-4<X≤0.001)、中组(0.001<X≤0.01)、大组(0.01<X≤0.1)和超扩增组(0.1<X≤1)。
共享克隆型评估
针对多样本项目,将进行受体库相似性评估以检测样本间克隆型共享水平。该分析在疾病组个体间克隆共享或治疗前后追踪研究中具有重要价值。采用Morisita重叠指数和直接重叠克隆型计数两种方法量化受体库相似度。
基因使用分析与谱型分型
通过评估受体库基因使用水平,解析不同受体库间的差异基因使用模式,观察相似性并确定基因组合对受体库多样性的贡献度。计算基因相关性矩阵和JS散度(Jensen-Shannon divergence)进行可视化。基于余弦相似度对每个基因的使用模式进行层次聚类。对于超过两个样本的项目,将基于基因使用进行降维,通过PCA和多维标度(MDS)图展示受体库间距离(JS散度用于预处理相似性)。同时进行谱型分型分析基因在各序列长度上的分布,计算并可视化高频基因片段。
受体库多样性评估
采用多种方法评估受体库多样性:Chao1估计器(评估物种丰富度)、Hill数(反映有效物种数)、真实多样性(有效克隆型数)、Gini-Simpson指数(随机两个克隆型相同的概率)、逆Simpson指数、D50值(覆盖50%总reads的最小克隆型数)以及稀疏分析(通过外推法评估物种丰富度)。
高频克隆型追踪
类似于共享克隆型评估,但聚焦于高频克隆型的跨受体库比较。该分析在群体水平比较或个体时序追踪等研究中具有应用价值。
多模态整合分析准备
对受体库对应的scRNA-seq样本进行读取和过滤(剔除表达基因数<200或>6000的细胞,该阈值针对10X Genomics数据设定)。过滤后数据经对数标准化和高变基因缩放,先进行PCA线性降维,再进行UMAP/t-SNE非线性降维。多样本将进行整合分析(根据用户选择采用seurat或Harmony方法),具体流程同scRNA-seq分析部分。整合后基于PCA或Harmony校正嵌入进行UMAP降维,使用前30个主成分通过k-NN/SNN和经典Louvain算法聚类,并通过自动化注释预测各簇细胞类型。
多模态整合分析
采用scRepertoire进行整合:基于细胞条形码将免疫受体库映射至对应scRNA-seq样本,通过标准克隆型定义、VDJC基因和CDR3核苷酸序列进行克隆鉴定。按前述五级分组评估克隆空间稳态,并将该信息可视化于scRNA-seq降维UMAP嵌入中。计算各预测细胞类型中克隆扩增组的比例,若元数据提供分组信息(如疾病组/对照组),将分别可视化组间比例差异。同时评估各细胞类型的克隆多样性,通过UMAP二维空间上的等高线密度图展示克隆扩增分布,并通过克隆网络分析展示细胞类型间共享克隆型的互作关系。
单细胞拷贝数变异(scCNV)分析框架
本分析流程针对10X Genomics多模态定量后的scCNV样本设计,支持多样本整合分析。由于CNV数据通常较其他组学数据量大,分析过程可能更为耗时。该工作流程主要基于10X Genomics官方scCNV应用演示文档构建(图6)。
质量控制与数据处理
针对每个样本,平台将读取包含拷贝数调用信息和基因组可映射区域的bed文件,并与样本指标摘要文件中的细胞身份信息进行交叉验证。在每个CNV位点,拷贝数信息将被归类为10个不同的ploidy等级(0-9),所有≥10的ploidy值归入同一等级。同时标记低质量细胞(由Cellranger识别的ploidy估计不确定且reads计数谱异常的细胞)。保留二倍体细胞中事件置信度>100的CNV事件,以及非二倍体细胞中事件置信度>50的CNV事件。基于样本中每个细胞的CNV谱进行层次聚类,将具有相似CNV事件的细胞归集,聚类结果将以可视化形式展示。同时计算并可视化每个细胞在各CNV事件中的平均ploidy值。
非二倍体细胞分析
筛选非二倍体细胞进行深入分析,保留可映射区域>2000kb且置信度>15的CNV事件。剔除在样本所有细胞中占比<5%的CNV事件。对最终CNV事件进行二值化(存在/不存在)后,采用判别分析主成分法(DAPC)进行聚类分析:基于降维后的PCA成分运行连续K均值算法,通过贝叶斯信息准则(BIC)计算拟合优度并自动确定最佳聚类数。对二值化CNV事件进行UMAP降维,可视化降维与聚类结果。通过对二值化非二倍体矩阵进行层次聚类,观察各细胞簇中明显的CNV簇模式。筛选非二倍体细胞中前50个CNV事件,展示其染色体位点的ploidy信息并按聚类结果分组。对于多样本项目,将评估并绘制各样本非二倍体细胞的簇占比,同时通过比较各簇的中位拷贝数构建系统发育树。
质谱流式(CyTOF)分析框架
本分析流程专为质谱流式细胞术(CyTOF)设计的.fcs文件分析而构建,支持多样本提交。经过去除双联体处理后的过滤.fcs文件将作为分析输入文件。
质量控制与数据处理
读取FCS格式文件后,对每个样本的表达矩阵进行弧正弦转换(co-factor=5)实现数据标准化。若提供批次信息,将采用线性回归校正批次效应。可视化各样本细胞计数概况,并通过各样本检测通道中标志物的密度分布进行质控验证。计算多维度标度(MDS)降维结果,展示样本间距离以评估组内和组间差异。计算各样本标志物的中位表达量,通过层次聚类热图展示所有样本的中位表达模式,同时提供样本箱线图。对于多组别项目,额外返回组别箱线图。计算各样本中标志物的非冗余评分,识别跨样本变异度最高的标志物。
整合降维与聚类分析
多样本项目在批次校正后合并进行降维分析。首先采用FlowSOM进行第一级降维与聚类,将细胞维度压缩至100个元簇(meta cluster),每个元簇包含标志物表达高度相似的细胞群。随后对降维矩阵采用共识聚类(CC)方法生成聚类结果,通过肘部法则(Elbow method)基于CC输出确定最终簇数。为二维可视化,先计算PCA嵌入再执行UMAP降维,将聚类结果映射至PCA和UMAP空间展示。FlowSOM生成的元簇也通过UMAP和t-SNE降维,用于评估元簇质量。
表型特征发现
聚类完成后,计算各簇标志物的中位表达量,通过热图和箱线图可视化。绘制各样本细胞中标志物的全局表达密度分布图,同时展示各簇的样本组成比例。通过计算各簇的表型特征预测细胞身份:将各簇标志物的中位表达量与整体数据中位值比较,筛选倍数变化>1.25的标志物作为该簇的特征标志物(如"簇1:CD3+CD4+"),最终生成各簇的表型标志物列表。
流式细胞术分析框架
本分析流程专为原始流式细胞术.fcs文件设计,支持多样本提交。
质量控制与门控策略
读取FCS格式文件后,对每个样本的表达矩阵进行弧正弦转换(co-factor=150)实现数据标准化。通过可视化各样本检测通道中标志物的密度分布进行质控验证。采用自动化门控策略去除碎片和双联体,具体步骤如下(均以去除碎片和双联体为目的):FSC-A vs SSC-A、FSC vs FSC-H、SSC-W vs SSC-H以及FSC-W和FSC-H的后续二元门控。每个门控步骤均提供可视化结果,并向用户提交门控策略汇总表。门控后重新评估各标志物的密度分布进行质控复核。与CyTOF分析类似,计算各样本标志物的中位表达量,通过层次聚类热图展示所有样本的中位表达模式。计算多维度标度(MDS)降维结果展示样本间距离以评估组内和组间差异,同时计算各样本中标志物的非冗余评分以识别高变异度标志物。
整合降维与聚类分析
多样本项目在样本合并后进行降维分析。类似CyTOF分析方法,首先采用FlowSOM将细胞元聚类为100个维度,每个元簇包含标志物表达高度相似的细胞群。随后对降维矩阵采用共识聚类(CC)方法生成聚类结果,通过肘部法则确定最终簇数。为二维可视化,先计算PCA嵌入再执行UMAP降维,将聚类结果映射至PCA和UMAP空间展示。
表型特征发现
聚类完成后,计算各簇标志物的中位表达量,通过热图和箱线图可视化。绘制各样本细胞中标志物的全局表达密度分布图,同时展示各簇的样本组成比例。
空间转录组分析框架
本分析流程针对10X Genomics定量后的空间转录组数据设计,可直接使用Cellranger的输出作为输入文件。支持多样本整合分析,同时提供多模态整合分析功能(目前仅限单样本多模态整合)。
质量控制指标评估与数据处理
使用seurat软件包对每个样本执行以下操作:计算每个spot的RNA分子数并通过小提琴图和空间成像数据可视化。采用sctransform方法(基于正则化负二项模型)进行标准化以消除技术变异。计算每个样本spots间的高变基因并在空间成像数据上可视化表达模式。
降维、聚类与差异表达分析
作为本质上的单细胞转录组数据,其降维聚类流程与scRNA-seq分析类似:对每个样本的sctransform转换数据执行PCA分析,基于前30个主成分进行UMAP投影。根据前30个PCA维度计算k近邻(k-NN)和共享近邻(SNN),通过原始Louvain算法进行模块化优化聚类。降维聚类结果既在UMAP嵌入空间展示,又映射至原始空间成像数据以呈现簇的空间分布特征。对每个簇进行差异表达分析,剔除在任一组细胞中检出比例<25%的基因,保留Bonferroni校正后p<0.1且平均log2倍数变化>0.25的差异基因(DEGs)用于辅助细胞类型注释。同时采用SingleR进行自动化细胞类型注释。
多样本分析
对于包含多个样本的项目,先合并样本再进行降维聚类,具体步骤同单样本分析。
多模态整合分析
当同时提供空间转录组和scRNA-seq数据时,分析流程类似于scImmune Profiling的多模态整合:读取scRNA-seq样本后,过滤表达基因数<200或>6000的细胞(当前参数针对10X Genomics数据优化)。过滤后数据采用与空间样本相同的sctransform方法标准化,经PCA(取前30个主成分)和UMAP降维后,基于前30个PCA维度通过k-NN/SNN和Louvain算法聚类。通过自动化注释预测scRNA-seq数据的细胞类型后,执行以下整合步骤:1)在空间数据与scRNA-seq数据间识别转移锚点细胞;2)将scRNA-seq预测的细胞类型标签转移至空间样本。最终可视化每个scRNA-seq预测细胞类型在空间各簇中的预测得分分布。
代码和数据可用性:
Single Cell Analyst在线版本访问地址:www.singlecellanalyst.org。该网络服务器的Docker版本及相应代码维护于GitHub仓库:https://github.com/singlecellanalyst/SCAWebserver。所用数据与脚本保存于GitHub:https://github.com/singlecellanalyst/SCAWebserver。平台使用的软件包列表详见文章附表3。SingleCellAnalyst平台工作流程演示所用数据集可从10X Genomics数据集网站获取:www.10xgenomics.com/datasets。补充材料(图表、图形摘要、幻灯片、视频、中文翻译版本及更新材料)可通过在线DOI或iMeta Science官网查阅:http://www.imeta.science/。
引文格式:
Lu Pan, Bufu Tang, Xuan Zhang, Paolo Parini, Roman Tremme, Joseph Loscalzo, Volker M. Lauschke, Bradley A. Maron, Paola Paci, Ingemar Ernberg, Nguan Soon Tan, Ákos Végvári, Zehuan Liao, Sundararaman Rengarajan, Roman Zubarev, Yuxuan Fan,Xu Zheng, Xinyue Jian, Ren Sheng, Zhenning Wang, Xuexin Li. 2025. “Comprehensive analysis of multi-omics single-cell data using the single cell analyst.” iMeta 4: e70038. https://doi.org/10.1002/imt2.70038
作者简介

潘璐(第一作者)
● 卡罗林斯卡医学院博士后。
● 研究方向为单细胞组学技术、鉴定肌萎缩侧索硬化症预后和诊断的生物标志物,近五年发表SCI10余篇,以第一作者发表于《Nature Communications》《Nuclei Acids Research》《Genome Biology》 等杂志,累计影响因子超过200分。

汤步富(第一作者)
● 复旦大学附属中山医院放疗科住院医师。
● 研究方向为肝癌的放射治疗和免疫微环境研究。参与国家重大专项、面上项目2项,参编专著1部。近五年来,发表SCI论文40余篇,包括《The Journal of Clinical Investigation》《PNAS》《Advanced Science》《Bioactive Materials》等期刊,以一作/通讯作者发表SCI论文20篇, IF共157分,其中top期刊6篇,H指数15。

张旋(第一作者)
● 博士,云南省高层次卫生健康技术医学后备人才,云南省肿瘤医院结直肠外科。
● 主要结直肠肿瘤的免疫治疗临床研究和免疫耐药基础研究工作。主持省级基础研究项目3项,临床研究2项。在JNCCN、BMC Medicine等期刊上发表SCI论文10余篇,IF>100分。研究成果多次被ASCO、KSMO及CSCO选为口头报告或壁报摘要。

王振宁(通讯作者)
● 主任医师、教授、博士研究生导师,胃肠肿瘤精准诊疗教育部重点实验室负责人。
● 教育部长江学者特聘教授,万人计划领军人才。获国家科技进步二等奖2项、省部级科技进步一等奖3项。研究方向为胃肠癌发生和转移的基础与临床研究。兼任中华医学会肿瘤学分会副主任委员、中国抗癌协会类器官与器官芯片专业委员会主任委员。

李薛鑫(通讯作者)
● 中国医科大学附属第四医院生物医学创新中心主任、“兴辽英才”青年拔尖人才、瑞典卡罗林斯卡医学院客座研究员。
● 主要研究方向:基于系统医学的泛癌多维解析与靶点挖掘及医学转化应用。近三年以通讯作者在Nature Communications、Genome Biology、PNAS、Cell Reports Medicine、 Hepatology等期刊发表论文多篇,主持瑞典卡罗林斯卡医学院国际合作项目 2 项、 国家自然科学基金面上项目及省部级项目 4 项。

盛韧(通讯作者)
● 教授,博士生导师,现就职于东北大学生命科学与健康学院,独立PI。
● 研究方向:细胞信号传导、肿瘤靶向治疗、肿瘤免疫微环境及类器官培养。近年来于Advanced Science、Cell Reports Medicine、Journal of Clinical Investigation、Hepatology、PNAS等杂志以通讯作者发表文章多篇。主持国家自然科学基金4项,省部级人才项目与基金9项。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期
iMetaOmics封面

1卷1期

1卷2期

2卷1期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,中科院分区生物学1区Top,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









