






FlexPose 应用于蛋白-小分子柔性对接场景下,能够在欧几里得空间中直接对蛋白-小分子复合结构的进行预测的等变神经网络模型,而无需传统的采样和评分策略。此模型考虑了蛋白氨基酸主链和侧链的柔性,会根据小分子的情况对氨基酸的侧链和主链进行调整,以实现柔性对接。
一、背景介绍
FlexPose 来源于中国深圳中山大学智能系统工程学院智能医疗中心的 Calvin Yu-Chian Chen 教授为通讯作者的文章:《Equivariant Flexible Modeling of the Protein−Ligand Binding Pose with Geometric Deep Learning》。文章链接:https://pubs.acs.org/doi/10.1021/acs.jctc.3c00273。该文章在 2023 年 11 月 8 日发表于 《Journal of Chemical Theory and Computation》 上。

蛋白质-配体复合物结构的柔性建模是计算机辅助药物开发中的一项基本挑战。近期的研究通过引入深度学习步骤改进了常用的对接工具。然而,这些策略由于保留了大量的采样且缺乏对生物分子柔性变化的考虑,限制了其精度和效率。在本项工作中,作者提出了 FlexPose,一个几何图网络,能够在欧几里得空间中直接进行复合结构的柔性建模,而无需传统的采样和评分策略。模型采用了两个关键设计:标量-向量双重特征表示和SE(3)等变网络,以应对动态结构变化。此外,作者还采用了两种策略:构象感知的预训练和弱监督学习,以增强模型在未知化学空间中的泛化能力。得益于这些范式,FlexPose 在涉及蛋白质构象变化的任务中大幅超越了所有测试过的流行对接工具和最新的深度学习方法。作者进一步通过两种构象感知策略探讨了蛋白质和配体相似性对模型性能的影响。此外,FlexPose 还提供亲和力估计和模型置信度以供后续分析。
二、模型介绍
了解小分子药物和靶标结合模式是理解小分子发挥生物活性的重要步骤。分子对接方法是传统实验方法的常用替代方案。它主要包含采样和评分两个阶段。采样阶段涉及在 3D 空间中搜索并提供构象(即结构)候选;评分阶段使用评分函数(如分子力场)对构象进行排序,最高分的构象被视为最终预测。然而,最近的研究表明,流行的对接工具在准确性(<60%)和蛋白质构象变化(<15%)的情况下表现依然有限。根据“诱导契合理论”,蛋白质上的配体结合口袋会响应配体的物理化学相互作用,在天然的游离构象(apo)和配体结合构象(holo)之间发生变化。不幸的是,由于自由度过多,对配体和蛋白质进行采样(即柔性对接)代价高昂。为节省时间,对接工具在实际应用(如高通量虚拟筛选)中通常将蛋白质视为刚性实体(半柔性对接,忽略蛋白质构象变化)。因此,复杂结构的精确且高速的柔性建模是传统工具面临的根本挑战。
随着人工智能的蓬勃发展,数据驱动的生物结构建模已成为一个热点方法,并在蛋白质/RNA 结构上展现出高精度。对于复杂结构,许多深度学习方法试图通过结合传统对接工具来提高建模精度。然而,基于对接的混合方法把大量精力用于优化评分函数或开发对接后的处理步骤,使得它们的上限在很大程度上受到对接工具采样能力的限制。此外,在面对巨大的采样空间(如使用游离态蛋白质进行柔性对接)时,这些模型的能力上限往往较低。
近期的研究使用端到端可微分的深度学习方法建模复杂结构,作为传统对接的潜在替代方案。Stärk 等人开发了 EquiBind,He 等人开发了 LigPose,分别用于无靶位对接(不指定结合位点)和局部对接(指定结合位点),它们均实现了高准确性和效率。此外,Corso 等人开发了 DiffDock,利用一种基于扩散的迭代过程从噪声分布生成结构。然而,这些方法将蛋白质视为刚性实体,忽略了蛋白质内部的动态结构变化建模。
为克服上述瓶颈,作者提出了一种全新的无对接深度学习框架——FlexPose,用于直接、柔性、端到端的复合结构建模,不遵循传统的对接范式(即采样和评分)。FlexPose 使用了两个设计来管理动态结构变化(标量-向量双特征表示和 SE(3) 等变网络)和两种策略来增强模型在未知化学空间中的泛化性(构象感知的预训练和构象感知的弱监督学习)。简而言之,FlexPose 将蛋白质-配体对表示为一个包含标量和向量特征的图(节点和边的集合)。与典型的分子图表示仅包含标量特征不同,附加的向量特征丰富了几何表示。然后,该图由编码器-解码器风格的网络处理,即 VAE 架构。两个构象感知的预训练编码器捕获蛋白质和配体的结构特征。基于预训练特征,解码器在欧几里得空间中直接预测并优化复合结构。它从零开始构建配体结构,而非像对接一样需要预生成的构象。模型框架是 SE(3) 等变的,使其对 3D 旋转和平移具有鲁棒性。此外,构象感知的弱监督学习使模型能够在数据稀缺的情况下,通过使用低置信度结构(如通过对接工具采样的结构)扩展学习到的化学空间。
在柔性建模任务中,FlexPose 在测试的所有对接工具和最近的基于深度学习的方法中表现出色,准确性比第二优方法高出最多 37.58% 。构象感知策略显著提高了模型在未知化学空间中结构的性能,即使在使用严格控制相似性的训练集训练后,FlexPose 的表现仍可与使用冗余数据集训练的其他深度学习方法媲美。此外,FlexPose 提供亲和力估计和模型置信度,在蛋白质-配体相互作用的后续分析中非常有用。进一步的消融研究证明了主要设计的有效性。该范式为蛋白质-配体复合物的柔性建模提供了新的视野,预计将为基于结构的药物开发奠定基础。
2.1 模型框架
下图是 FlexPose 的整体框架图。与大多数方法(如基于采样和评分的传统对接方法及对接混合深度学习方法)不同,FlexPose 能够对蛋白质-配体复合物结构进行端到端的柔性建模(图 a),估计亲和力和模型置信度,而不受输入蛋白质构象(apo 或 holo)的限制。换言之,FlexPose 可以输入蛋白的 Apo 或者 Holo结构以及小分子的 2D 结构,预测小分子的结合 Pose 以及 氨基酸的侧链变化情况。
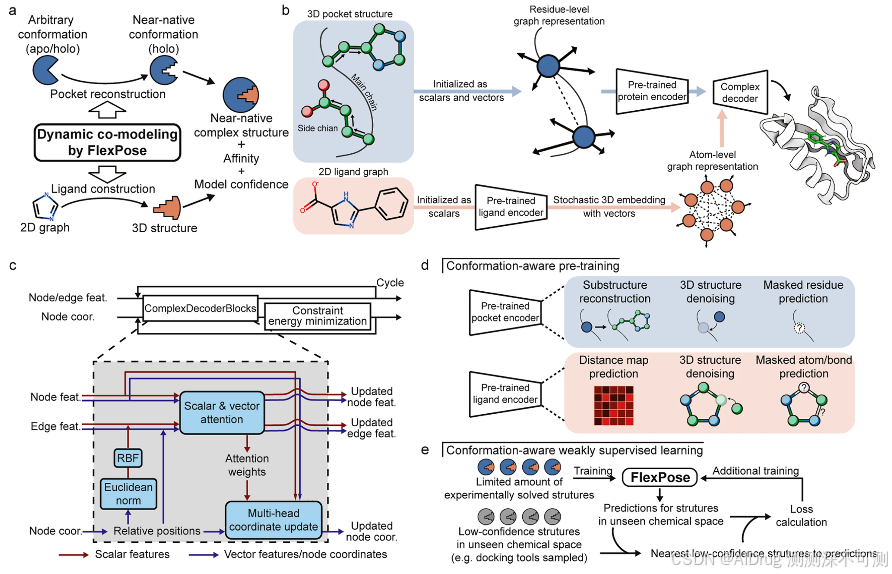
FlexPose 将蛋白质-配体对表示为一个包含节点(蛋白质为残基级别、配体为原子级别)和边的图(图 b),其中每个节点和边都初始化为标量(生化特性,如残基/原子类型)和一组欧几里得向量(几何特征,如蛋白质原子间的相对位置,例如:CA-CB)。不同于通常只包含标量特征的方法(通常仅使用距离或扭转角表示几何特征),这种标量-向量双重特征表示使得 FlexPose 能够显式管理节点的动态变化,而不仅仅依赖与所有相邻节点的相对位置。此外,双重特征允许 FlexPose 在无需直接访问其原子结构的情况下,涵盖更多的蛋白质残基并提供充分的几何表示。
FlexPose 采用编码器-解码器的设计风格。蛋白质编码器和配体编码器首先将 3D 蛋白质结构和 2D 配体图分别嵌入到潜在空间中。然后,复合物解码器从零开始构建配体结构(小分子从随机 3D 嵌入,即随机高斯噪声开始)并循环更新,同时重建蛋白质结构。在每个循环中,复合物解码器迭代地更新特征和结构。简单来说,复合物解码器通过消息传递来处理标量和向量特征,其中每个节点通过聚合来自其相邻节点的加权消息(由注意力机制提供权重)来更新其特征。需要注意的是,注意力权重用于结构和特征的更新。在每个循环的末尾,复合物解码器对配体执行约束能量最小化,以确保几何结构的正确性(这一点在其他模型中很少见)。然后,将特征和结构发送到下一个循环以进行进一步优化,如图 c 所示。附加的浅层网络用于利用更新的特征估计亲和力。
为了提高模型的泛化能力,作者使用构象感知任务对编码器进行预训练,并提出了基于构象的 WSL(弱监督学习)以扩展学习的化学空间。构象感知预训练通过与化学性质相关的任务(如掩码属性预测)学习标量特征,并通过与 3D 结构相关的任务(如结构去噪)学习向量特征,从而使解码器能够构建具有更好特征表示的结构(图 d )。对于构象感知 WSL,它鼓励未见结构(例如实验上未解的结构)尽可能接近最近的低置信度结构(即计算得到的结构)(图 e)。为减少偏差,优选通过自下而上的方法生成低置信度复合物结构,例如量子力学/分子力学等方法。这有助于在预测的化学空间与训练样本几乎完全不同的情况下,研究新颖的结构。
为了进行更全面的评估,作者构建了一系列具有不同设置的变体(如 、
、
,如下表)。主要差别在,是否使用预训练的 encoder,是否使用 apo 的结构进行训练、是否做了能量对消化限制、以及构象感知的弱监督学习(WSL)。

标准版本(即 FlexPose)及其所有变体均未使用构象感知 WSL,因为这是一项需要低置信度结构的计算密集型策略。构象感知 WSL 仅在考察训练集与测试集之间相似性的影响时进行评估。
2.2 数据集
本研究使用了 PDBbind 数据集(2020 版本)和 APObind 数据集。PDBbind 数据集包含 19,443 对天然蛋白质-配体复合物的配对数据,包括其结构和结合亲和力(以抑制常数(Ki)或解离常数(Kd)的对数变换形式表示)。该数据集有两个子集,即精炼集(N = 5316)和核心集(N = 285),具有更高的数据质量。由于 PDBbind 数据集仅提供蛋白质结构的配体结合构象(holo),因此作者引入了 APObind 数据集,后者收集了 PDBbind 数据集中 holo 结构对应的天然未结合构象(apo)。在本研究中,仅考虑 TM-score > 0.3 且骨架 RMSD < 15.0 Å 的 apo 结构。最终,在研究中使用了 9399 个 apo 结构。其蛋白质口袋定义为至少一个原子在任一配体重原子 10 Å 范围内的残基。
为评估模型,作者使用了三个新的测试集:apo-core 集、apo-refined 集和 cross-core 集。它们基于 PDBbind 数据集的核心集和精炼集构建,后者被广泛用于评估。所有在核心集中且在 APObind 数据集中有相应 apo 结构的配对数据被收集为 apo-core 集(178 对)。它用于基于 apo 蛋白质评估近天然复合结构(包括配体和 holo 蛋白质结构)的预测。然而,apo-core 集是一个相对较小的子集。作者进一步在精炼集中随机采样了 1000 对符合一些筛选条件(口袋中无间隙或缺失残基,TM-score > 0.8,骨架 RMSD < 4 Å)且具有相应 apo 蛋白结构的配对数据,构成了 apo-refined 集。cross-core 集是基于核心集构建的。核心集中的配对数据首先按蛋白质序列相似性聚类为 57 组,每组包含约 5 对。然后,在每个簇中,所有蛋白质与所有配体进行配对,生成了 1343 对 cross-core 集的数据。没有成功处理的数据未包含在评估中。最终,apo-core 集包含 168 对,apo-refined 集包含 937 对,cross-core 集包含 1305 对,核心集包含 278 对。对于 apo-core 集、cross-core 集和核心集,FlexPose 的标准版本使用了 9221 个 apo 结构和 18,781 个 holo 结构进行训练。对于 apo-refined 集,模型使用了 8462 个 apo 结构和 18,122 个 holo 结构进行训练。
作者在预训练阶段使用了两个较大的数据集:蛋白质口袋集和小有机分子(SOM)集。口袋集由 PDB 数据库构建。所有从 PDB 数据库收集的蛋白质通过 Fpocket 分析以提取其口袋。在筛选(可成药性得分 >0,得分 >0.01)后,共有 1,048,575 个口袋用于预训练。蛋白质口袋定义为至少一个原子在预测口袋中心 10 Å 范围内的残基。SOM 集包含来自 DrugBank 的 11,290 个 SOM,来自 ChEMBL 的 2,136,187 个 SOM,以及从 ZINC 中采样的 3,852,523 个 SOM。所有蛋白质通过 modeler 修复缺失原子,所有 SOM 通过 RDKit 处理。
2.3 模型性能
作者在 apo/polo 数据集上分别比较了 FlexPose 和其他现有方法的性能表现,并探究了结构感知的预训练和 WSL 对模型泛化能力的影响。作者还探究了结合亲和力和模型置信度对相互作用改进的分析。最后,作者通过消融实验探究了模型中关键设计的影响。
2.3.1 FlexPose 使用 Apo 蛋白质预测配体结合构象的表现
作者首先评估了 FlexPose 在使用 apo 蛋白质预测天然复合物结构的能力,这对传统的对接方法来说非常具有挑战性。大多数 holo 构象相较于 apo 构象显示出结构差异,通常是由于侧链构象的变化所致。传统的对接方法在处理结构变化时面临严重的采样压力,而且许多对接工具仅将蛋白质视为刚性组件(半柔性)。作者使用了两个测试集,这两个测试集分别来自 APObind 和 PDBbind 数据集,用于评估,分别命名为 apo-refined 集(N = 937)和 apo-core 集(N = 168)(它们包含天然复合物结构及相应的 apo 结构)。
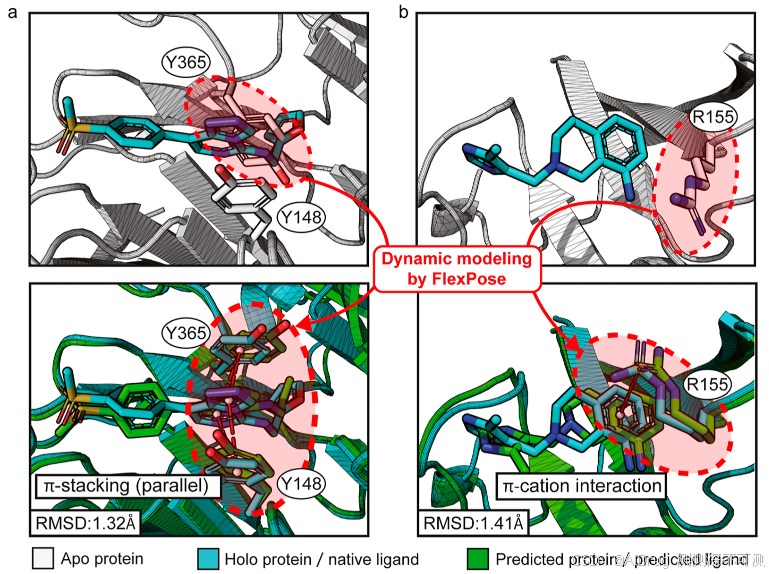
根据评估结果显示,FlexPose 显著优于所有测试的传统对接工具,在两个测试集中,预测构象与天然构象之间的均方根偏差(RMSD)值较小。下图 a 和 b 展示了两个样本在 apo/holo 状态下的构象(apo/holo PDB IDs: 2QXV/5WUK, 2JCP/4NP3)
下图 c 和 e 中显示了两个测试集的成功率(对接工具的 top-1 成功率)。成功率定义为 RMSD < 2 Å 的预测比例,这是先前研究建议的黄金标准。图 d 和 f 是在两个测试集上的累积 RMSD 分布情况。FlexPose 在 apo-refined 和 apo-core 集中的成功率分别为 56.24% 和 64.88%,比表现最好的对接工具(RosettaLigand)分别提高了 36.60% 和 40.48%。两个半柔性对接工具 Smina^{semi}(Smina 的半柔性设置)和 LeDock 在 apo-refined 和 apo-core 集上的成功率仅为 8.32%/8.33% 和 4.38%/2.98%,原因在于未处理蛋白质构象变化。两个柔性对接工具的表现优于半柔性对接工具,成功率分别为 8.22%/8.33%(Smina^{flex},即 Smina 的柔性设置)和 19.64%/23.21%(RosettaLigand)。然而,这些柔性对接工具需要探索侧链构象的大量采样空间,因此非常耗时。尽管 RosettaLigand 和Smina^{flex}在采样上投入了大量精力,但它们的成功率远低于 FlexPose(36.60%-56.55%)。总之,即使一些工具考虑了构象变化,所有测试的对接工具在此任务中均表现出瓶颈,FlexPose 明显优于它们。作者进一步将 FlexPose 与最近建立的先进深度学习方法进行比较,结果显示 FlexPose 取得了压倒性改进(成功率比第二好的深度学习方法提高了 33.41%)。大多数先前的深度学习方法旨在通过混合方式提高局部对接性能。作者选择了五种先进的深度学习方法进行比较:3 个评分函数(DeepDock、RTMscore 和 GNINA),1 个结构后精炼方法(MedusaGraph),和 1 个交互距离图预测器(EDM-Dock)。所有方法的成功率均在 apo-core 集上进行评估。RTMscore、DeepDock 和 MedusaGraph 通过 RosettaLigand 采样的结构进行评估。FlexPose(64.88%)表现最佳,其次是 EDM-Dock(31.47%)、RTMscore(24.40%)和 GNINA(21.82%)。DeepDock 和 MedusaGraph 显示出有限的能力(<2.38%)。因此,FlexPose 相较于其他测试的深度学习方法具有明显优势。EDM-Dock 和 RTMscore 相较于表现最好的对接工具(RosettaLigand)分别提高了 8.26% 和 1.19%,可能得益于其残基级表示。值得注意的是,对于这些高级评分函数,其上限受对接工具采样能力的限制(图 c 中的虚线)。对于表现最好的对接工具(RosettaLigand),仅有 55.36% 的蛋白质-配体对具有 RMSD < 2 Å 的候选姿势(这一比例甚至低于 FlexPose 的成功率,两者相差 9.52%)。因此,单纯优化评分函数很难提高其余蛋白质-配体对的准确性,这表明提出的框架突破了传统建模过程中的部分瓶颈。
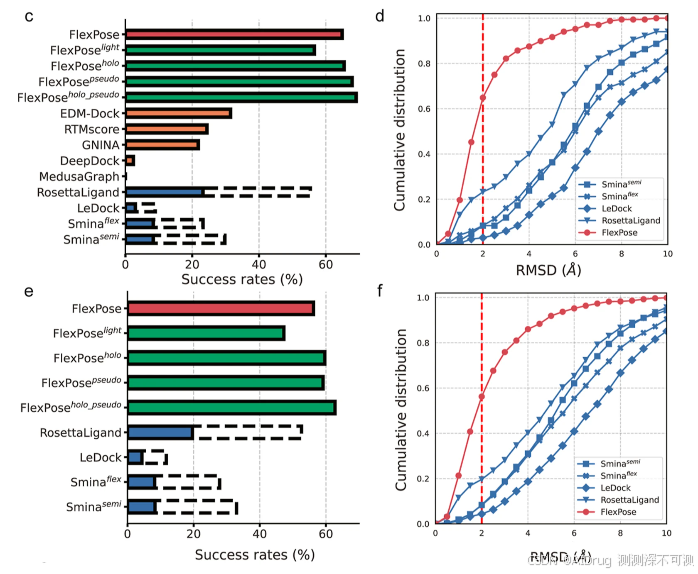
作者报告了四种 FlexPose 变体:(无编码器,模型尺寸较小)、
(仅用 holo 结构训练,无法预测蛋白质结构变化)、
(无能量最小化),以及
(结合
和
)。即便是
,与 RosettaLigand 相比,成功率在 apo-refined 和 apo-core 集上分别提高了 27.64% 和 33.34%。FlexPose 的 holo 和 pseudo 变体的表现略优于标准版本。尽管由于其设置原因无法保证生成物理合理的结构(例如可能包含原子冲突),但它们比 FlexPose 获得了更高的成功率,最高提高 6.51%,进一步表明了这种范式的潜力。
2.3.2 FlexPose 使用 Holo 蛋白质预测配体结合构象的表现
作者测试了 FlexPose 在 holo 蛋白质下预测配体结合构象的能力,涉及两种任务类型:重新对接和交叉对接。重新对接旨在重现使用该特定配体确定的 holo 蛋白质构象的天然复合物结构。交叉对接旨在重现来自不同配体的次优 holo 蛋白质构象。作者将 FlexPose 与 14 种传统或机器/深度学习方法进行了比较。评估基于从 PDBbind 数据集中提取的 core(N = 278)和 cross-core(N = 1305)集合,分别用于重新对接和交叉对接任务。
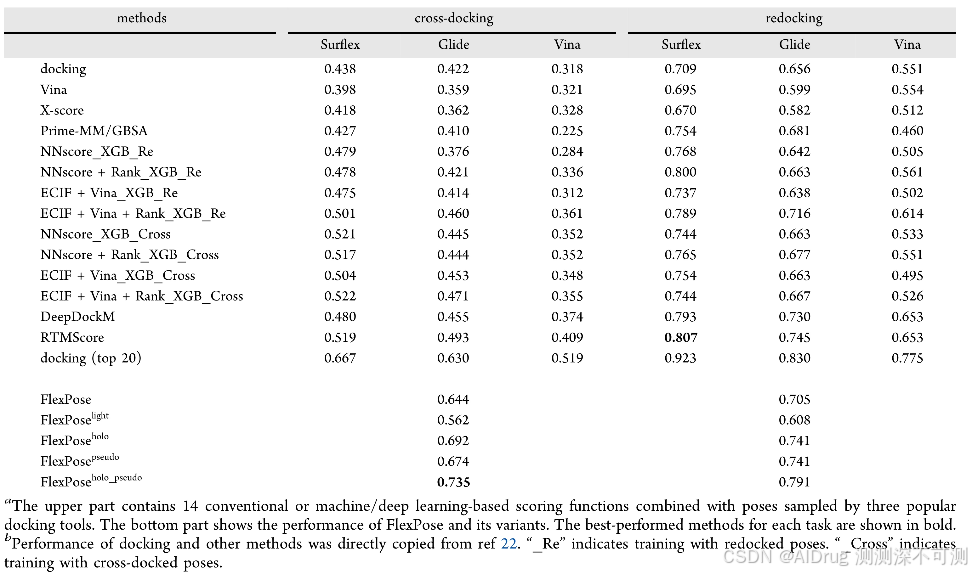
如下表所示,FlexPose 在交叉对接任务上显著优于所有测试的方法,并在重新对接任务上表现出可比的准确性。在交叉对接方面,FlexPose 及其变体的成功率在 0.644 到 0.735 之间,远高于第二好的方法 RTMscore(0.519)。在 cross-core 集合中的所有蛋白质-配体对中,只有 0.667 至少有一个采样姿势的 RMSD < 2 Å 值。值得注意的是,这一比例接近或低于 FlexPose 及其变体的成功率,表明了方法的有效性。在重新对接方面,FlexPose 及其变体的成功率在 0.705 到 0.791 之间,略低于表现最好的方法 RTMscore(0.807)。然而,交叉对接比重新对接更接近实际应用场景,因为重新对接在真实应用中几乎不会出现。结果表明,即便使用 holo 蛋白质,所有测试的对接工具和混合深度学习方法在准确性上表现仍有限,而 FlexPose 达到了更高的准确度。
2.3.3 结构感知的预训练和 WSL 提升了模型的泛化能力
作者在交叉对接任务中测试了使用不同分子相似性阈值训练的模型,同时比较了结构感知预训练和 WSL 的效率。训练集根据其与测试集的蛋白质和配体相似性进行了筛选。
作者使用 holo 结构训练了轻量化版本(称为 FlexPose^{holo\_light} )作为基线模型,并与添加了预训练编码器或 WSL 的相同模型进行了比较。对于预训练,使用了额外的蛋白质和小有机分子,通过结构感知任务训练两个编码器。预训练过程中未使用复合物结构。相比之下,WSL 使用低置信度的复合物结构在未见化学空间中对解码器进行了额外训练,损失权重较弱。WSL 计算预测结构与其最近的低置信度结构(即与预测结构 RMSD 最小的结构)之间的损失。在本研究中,作者采用了重新对接结构集(在核心集上),用于 WSL(每个复合物包含最多 10 Å RMSD 的 100 个结构,其中 13.30% 的结构 RMSD 小于 2 Å)。
如下图所示,相似性对性能有影响,且在添加了预训练编码器或 WSL 后,模型在低相似性阈值下表现显著提高。作者得出两个主要结论:(1)蛋白质相似性对模型性能的影响大于配体相似性。基线模型(即 FlexPose^{holo\_light},图 b)的成功率从配体相似性阈值 1.0 时的 0.59 降至 0.4 时的 0.39。然而,当蛋白质相似性阈值降至 0.4 时,模型成功率降至 0.20。这表明模型善于推断已知蛋白质的新结合模式,但在预测未见蛋白质时存在挑战,即使配体与训练集相似。这符合直觉,即蛋白质比配体更大,能提供比配体更多的学习交互模式。(2)预训练编码器和 WSL 都能显著提升模型性能(图 c,d),且 WSL 效果更佳。如图 a−d 所示,两者在大多数相似性阈值下均可提高成功率(最多提高 0.14)。训练集越严格(即低相似性阈值),提升效果越显著。例如,在蛋白质和配体的 1.0 相似性阈值下,预训练编码器和 WSL 分别相比基线提升了 0.02 和 0.04;在蛋白质和配体的 0.4 相似性阈值下,提升了 0.06 和 0.14。在大多数相似性阈值下,WSL 增强的模型优于仅使用预训练编码器的模型,表明低置信度结构的先验知识对于提升深度学习模型具有重要作用。
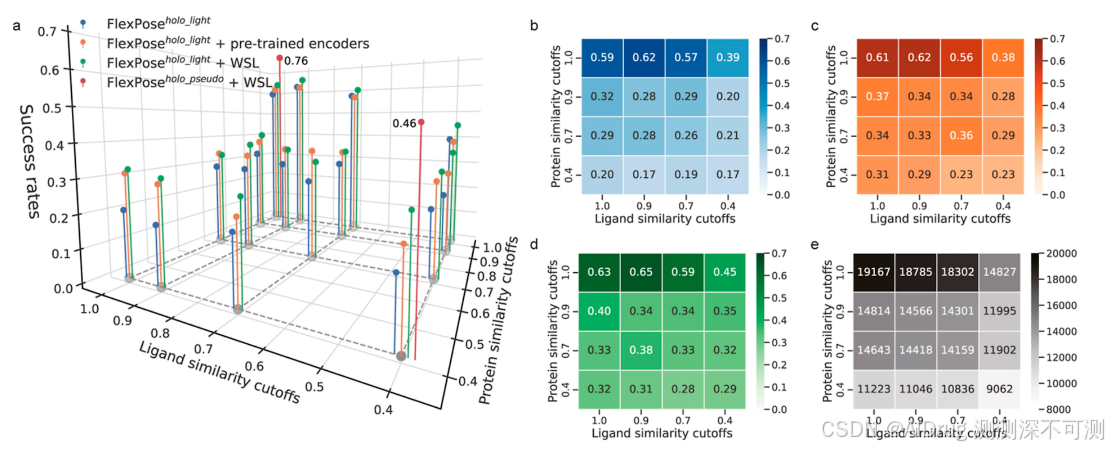
由于 是为全面研究相似性影响而构建的,作者进一步构建了两个带有 WSL 的
(更多参数),使用蛋白质和配体相似性阈值为 1.0 和 0.4 的数据集进行训练,以与先进方法进行比较。即使在 0.4 的相似性阈值下,
(成功率 0.46)也优于所有测试的对接工具(成功率 0.32−0.44),并且其准确度接近 RTMscore(成功率 0.52)。值得注意的是,RTMscore 是使用冗余数据集训练的(即没有相似性阈值的通用集)。在 1.0 相似性阈值下,
(成功率 0.76)显著优于所有对接工具和 RTMscore。这些结果强有力地证明了通过结构感知策略增强的模型在泛化能力方面的卓越表现。
2.3.4 高效的构象变化重建
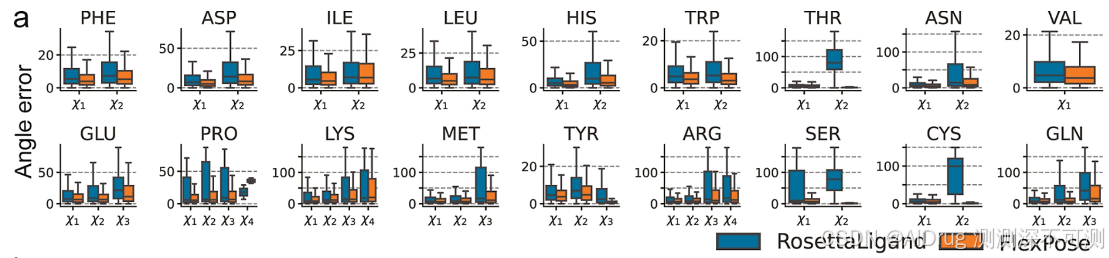
除了配体预测外,FlexPose 还能够准确预测蛋白质构象变化。它可以重建蛋白质的侧链,并提供骨架 CA 和 CB 原子的位置信息。如下图所示,对于所有氨基酸类型,FlexPose 在预测的侧链构象中获得了比 RosettaLigand 更低的 χ 角度误差。在 2.3.1 节的示例中,FlexPose 在从 apo 到 holo 构象的转变中重建了参与相互作用的残基。其中的红色圆圈揭示了 apo Y365 与原生配体构象之间的冲突,该冲突在预测中得到了改善。此外,FlexPose 的预测中建立了非共价相互作用(π-堆积和π-阳离子相互作用)(通过 PLIP 识别)。在骨架 CA 原子预测方面,FlexPose 在 apo-refined 和 apo-core 集上分别达到了 0.78 和 1.41 Å 的平均误差。以上结果表明,FlexPose 在重建蛋白质构象变化方面具有显著的精度。
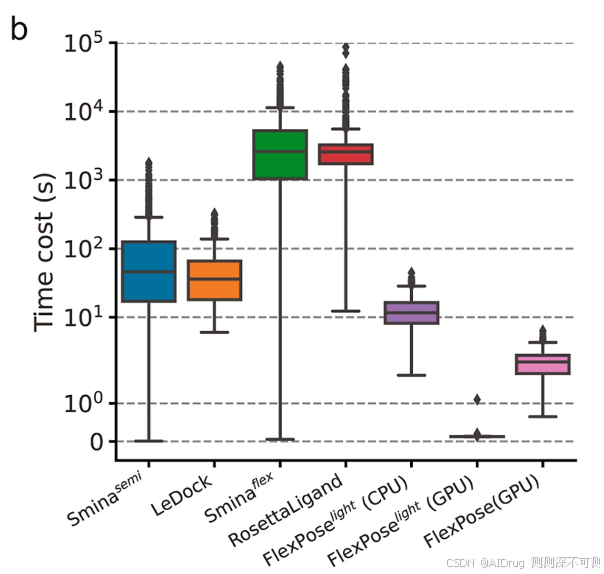
此外,FlexPose 展现出比传统对接工具更高的效率,如下图所示。在单个 CPU 核心(Intel(R) Xeon(R) Platinum 8255C)上,两个半柔性对接工具,Smina semi 和 LeDock,具有可接受的时间成本但精度有限(每次预测分别为 1.17 × 10^{2} 和 5.27 × 10^{1} 秒),分别比 长 8.86 和 3.99 倍。如果考虑蛋白质变化(即柔性对接),对接工具的效率则更低。两个柔性对接工具,
和 RosettaLigand,每次预测的平均时间分别为 4.32 × 10^3 秒和 3.66 × 10^3 秒,比
分别慢了 327 倍和 277 倍。使用 RTX 3090 GPU 设备时,FlexPose 比
和 RosettaLigand 快 1823 倍和 1544 倍。如此高的效率对于一些耗时的任务至关重要,例如药物筛选。
2.3.5 通过亲和力和模型置信度估计改进相互作用分析
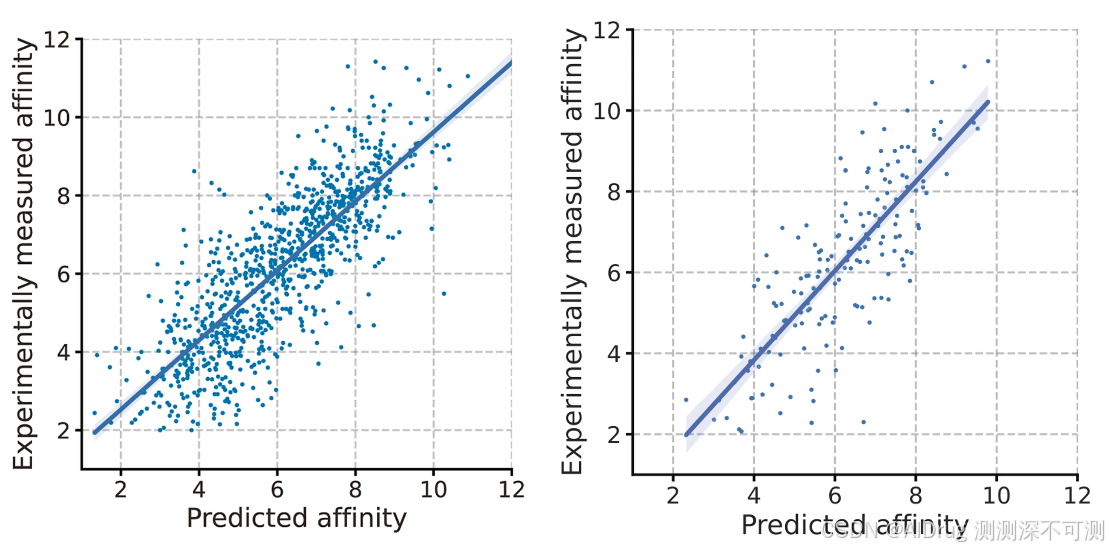
FlexPose 提供了亲和力和模型置信度的估计,这对于蛋白质-配体相互作用的后期分析非常有用。如下图所示,FlexPose 在 apo-refined 和 apo-core 集上分别达到了 0.809 和 0.801 的 Pearson 相关系数(Pearson R)用于亲和力估计。与其他基于结构的亲和力估计方法不同,FlexPose 在预测亲和力时不需要原生复合物结构,因为该结构已经包含在预测中。许多方法使用对接工具获得的结构作为替代品,但这些结构的表现通常不如原生结构。 这些发现证明了 FlexPose 多任务设计的高效性,能够提供全面的结构-活性关系学习。
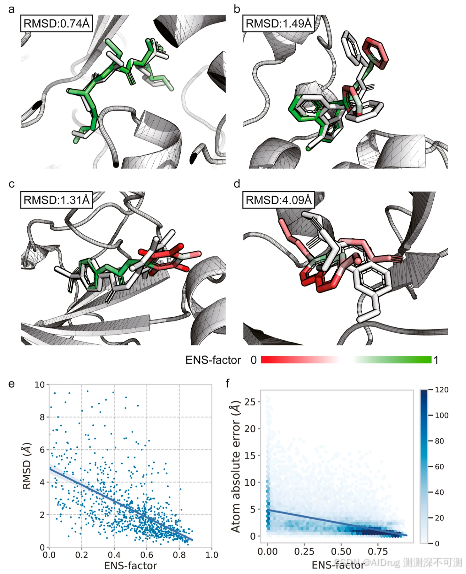
为了评估预测的可靠性,作者引入了一种名为“ENS 因子”的模型置信度指标,该指标不需要额外的可训练参数。它基于这样一个假设:FlexPose 更倾向于对置信部分产生一致的预测,而不考虑不同的初始 3D 嵌入。ENS 因子的值是通过简化表示预测结构集内的偏差来计算的。较高的 ENS 因子值表示高模型置信度。下图 a-d 展示了四个可视化样本。具有较高 ENS 因子值的子结构(即高置信度,显示为绿色)接近原生位置。相反,具有较低 ENS 因子值的子结构(即低置信度,显示为红色)表现出较大的误差。FlexPose 在 apo-refined 集上达到了分子级 ENS 因子与 rmsd 之间的 Pearson R 为 −0.61(图 e),在原子级 ENS 因子与原子位置误差之间的 Pearson R 为 −0.54(图 f)。这些结果表明,ENS 因子可以代表预测中的潜在误差,为进一步的结构分析(例如非共价相互作用)提供了直接的视觉参考。
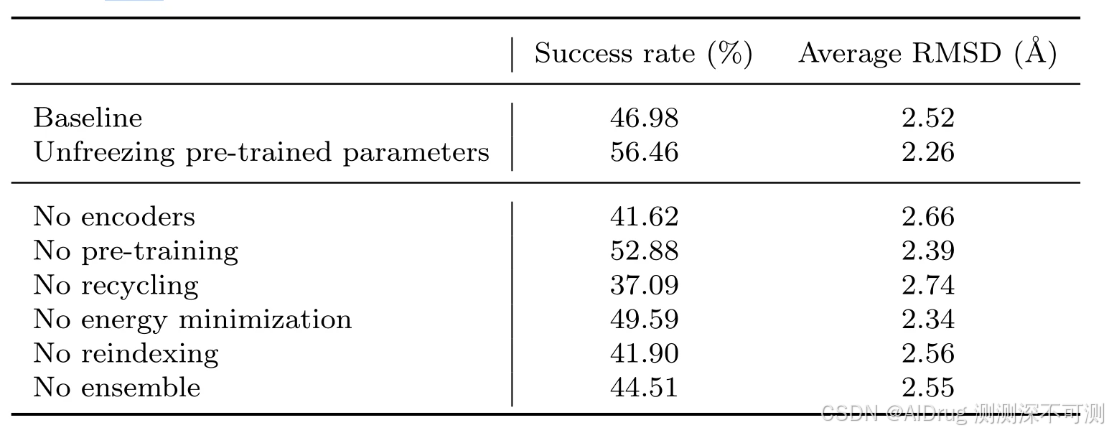
2.3.6 FlexPose的消融分析
作者进行了消融研究,以展示下表中列出的关键设计(包括架构和训练策略)的影响。没有预训练时,准确率下降了 3.58% 到 5.90%,表明构象感知预训练的效率。去除集成和回收策略后,准确率分别下降了 2.47% 和 9.89%。两个单独的独立预测之间的波动通过集成策略得到了稳定。回收策略则在不增加额外参数的情况下优化了预测,并大幅提高了准确性。这些结果表明,这些设计提高了模型的性能。
三、FlexPose 评测
3.1 安装环境
复制代码项目:
git clone https://github.com/tiejundong/FlexPose.git创建 FlexPose 环境并安装项目运行所需的一些基础的依赖库
# activate environment
conda create -n FlexPose python=3.8
conda activate FlexPose
# rdkit
conda install -c conda-forge rdkit
# torch
pip install torch==1.10.2+cu113 -f https://download.pytorch.org/whl/torch_stable.html
# torch_geometric
pip install torch_scatter==2.0.9 torch_sparse==0.6.13 torch_cluster==1.6.0 torch_spline_conv==1.2.1 -f https://data.pyg.org/whl/torch-1.10.2+cu113.html
pip install torch-geometric==2.0.4
# other
pip install ray keras_progbar
conda install scipy tqdm seaborn pandas ipykernel
conda install jupyterlab tensorboard einops biopandas chardet通过下面命令安装 modeller
# modeller
conda install modeller -c salilab上面安装的 modeller 需要许可证密钥激活才能使用,许可证密钥仅对学术人员免费提供,所以请使用教育邮箱注册申请。注册的网页链接是:Registration![]() https://salilab.org/modeller/registration.html ,页面截图如下。选择使用的平台时建议全选,这样申请的许可证密钥可以跨平台使用。个人信息填写好之后,提交注册申请,系统会给注册的邮箱发送许可证密钥(可使用学校邮箱注册获取的许可许可证密钥)。
https://salilab.org/modeller/registration.html ,页面截图如下。选择使用的平台时建议全选,这样申请的许可证密钥可以跨平台使用。个人信息填写好之后,提交注册申请,系统会给注册的邮箱发送许可证密钥(可使用学校邮箱注册获取的许可许可证密钥)。
通过 conda 安装好 modeller 后,打开 FlexPose 环境下 modeller 的配置文件 /workspace/anaconda3/envs/FlexPose/lib/modeller-10.6/modlib/modeller/config.py,内容如下:
install_dir = r'/workspace/anaconda3/envs/FlexPose/lib/modeller-10.6'
license = r'XXXX'把 license 替换为申请的许可证密钥,即修改成:
install_dir = r'/workspace/anaconda3/envs/FlexPose/lib/modeller-10.6'
license = r'xxxxxx' # 自行替换根据说明文档,使用下面的命令安装 pyrosetta 。
# pyrosetta
# username/password are provided with when you obtained a Rosetta license
conda install -c https://username:password@conda.graylab.jhu.edu pyrosetta
3.2 对接构象生成案例测试
3.2.1 内置案例
项目提供内置的测试案例是 4r6e.pdb 蛋白,配体 3JD 在口袋中的构象如下:
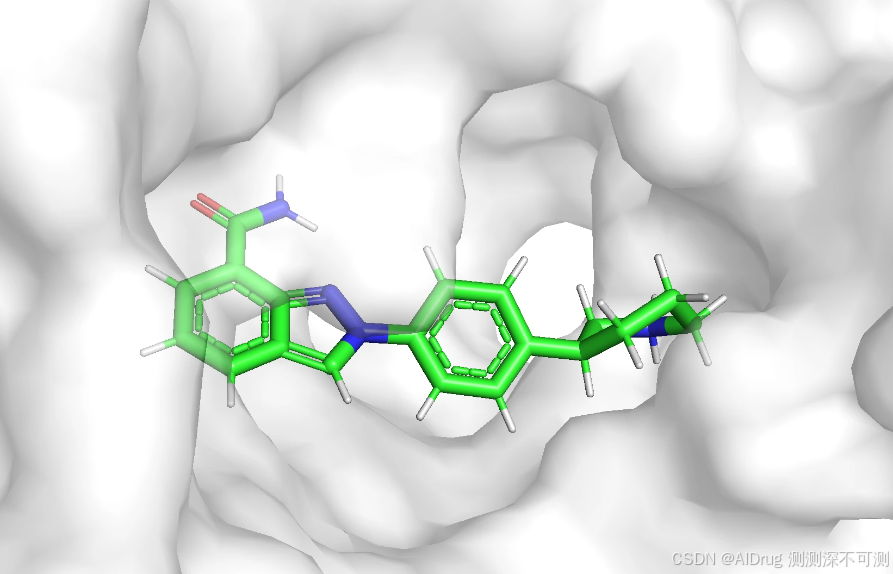
配体的 2D 结构如下:
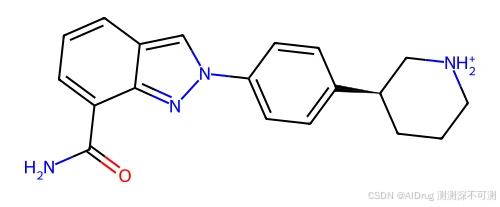
项目提供处理好的输入数据,保存在 ./FlexPose/example/4r6e 文件夹中,其中的文件如下所示:
.
|-- 4r6e_ligand.mol2
`-- 4r6e_protein.pdb
0 directories, 2 files项目提供的 ./FlexPose/utils/prediction.py 脚本中 predict 函数可以用来预测配体在口袋中的对接构象,函数的参数如下所示。
def predict(
param_path=None, # model parameter path
protein=None, # protein path
ligand=None, # ligand path
ref_pocket_center=None, # ligand-like file path
batch_csv=None, # batch prediction
model_conf=False, # predict model confidence
device='cuda:0',
ens=1, # ensemble number
seed=42,
batch_size=12, # only work with ens=1
num_workers=4, # only work with ens=1
prepare_data_with_multi_cpu=False, # prepare inputs with multi-processing
min=True, # torch build-in minimization
min_type='GD', # ['GD', 'LBFGS']
rdkit_min=False, # rdkit minimization
env_min=False,
cache_path='./cache',
output_structure=True,
save_mol=False,
save_only_pocket=False,
structure_output_path='./structure_output',
output_result_path=None,
calc_rmsd=False,
min_loop=100, # minimization loop
min_constraint=5, # minimization constraint factor
):
...其中,protein 和 ligand 用来指定蛋白和原始配体的结构文件,蛋白结构是 pdb 格式的文件,而 ligand 可以指定 sdf 、pdb、mol 和 mol2 格式的文件,也可以指定分子的 SMILES。如果读取的是分子 SMILES,首先会使用 AllChem.EmbedMolecule 来为该分子生成一个初始的三维构象。接着,使用 MMFF(Merck Molecular Force Field)对分子进行能量优化。优化时,原子的最大位移限制为 1 Å。最后,执行 20 次最大化步骤的能量最小化。至此从 SMILES 得到分子构象。根据 RDKit 生成的构象再预测对接到蛋白口袋中的构象。ref_pocket_center 用来指定参考配体通过 Fpocket 来选择蛋白口袋。model_conf 设置是否预测对接构象模型的置信度。为了复现预测结果,默认设置参数 seed=42 。structure_output_path 指定输出结果的保存路径。
4r6e_ligand.mol2 和 4r6e_protein.pdb 是配体和蛋白的结构文件,作为模型的输入。创建demo.ipynb 来使用 FlexPose 模型预测蛋白-配体的对接构象和结合亲和力。预测分别输入配体和蛋白结构文件,把配体结构的几何中心作为对接口袋中心。输出的配体和蛋白对接构象、预测的结合亲和力等信息保存在 ./4R6E 文件夹中。模型预测对接构象设置默认随机数(seed=42),我们设置三个不同的随机数(42, 425, 2024),然后检查三个不同的随机数下生成的 pose 是否相同,具体代码如下:
from FlexPose.utils.prediction import predict as predict_by_FlexPose
seed_list = [42, 425, 2024]
for seed in seed_list:
predict_by_FlexPose(
protein='./FlexPose/example/4r6e/4r6e_protein.pdb',
ligand='./FlexPose/example/4r6e/4r6e_ligand.mol2',
ref_pocket_center='./FlexPose/example/4r6e/4r6e_ligand.mol2',
model_conf=True,
seed=seed,
ens=10,
device='cuda:0',
structure_output_path=f'./4R6E/structure_output_{seed}',
output_result_path=f'./4R6E/output_{seed}.csv',
)代码运行输出:
==================== Running FlexPose ====================
Preparing input data: 0%| | 0/1 [00:00<?, ?it/s]
MODELLER 10.6, 2024/10/17, r12888
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2024 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, G.Q. Dong,
M.A. Marti-Renom, N. Eswar, F. Alber, M. Topf, B. Oliva,
A. Fiser, R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux 7ab82433c90d655e 6.5.0-35-generic x86_64
Date and time of compilation : 2024/10/17 20:34:40
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2024/11/11 10:20:31
read_to_681_> topology.submodel read from topology file: 3
core.io.rcsb.ExperimentalTechnique: [ ERROR ] Unrecognized experimental technique string 'THEORETICAL MODEL, MODELLER 10.6 2024/11/11 10:20:31'
Prepared data: 1/1, 100.00%
Loading FlexPose parameters ...
Downloading: "http://www.knightofnight.com/upload/data/FlexPose/FlexPose_param.chk" to /home/wufeil/.cache/torch/hub/checkpoints/FlexPose_param.chk
...根据输出结果可以看出,成功导入 modeller 。第一次运行上面的代码会自动下载项目提供的训练好的模型到 /home/wufeil/.cache/torch/hub/checkpoints/FlexPose_param.chk 。模型的下载链接为:http://www.knightofnight.com/upload/data/FlexPose/FlexPose_param.chk ,如果服务器下载较慢,可以使用浏览器访问下载,把模型上传到对应文件夹即可。
模型输出结果到 ./4R6E 文件夹中,目录结构如下。三个随机数后缀的文件夹和 csv 表格分别保存对接构象和预测的结合亲和力等信息。
.
|-- output_2024.csv
|-- output_42.csv
|-- output_425.csv
|-- structure_output_2024
|-- structure_output_42
`-- structure_output_425
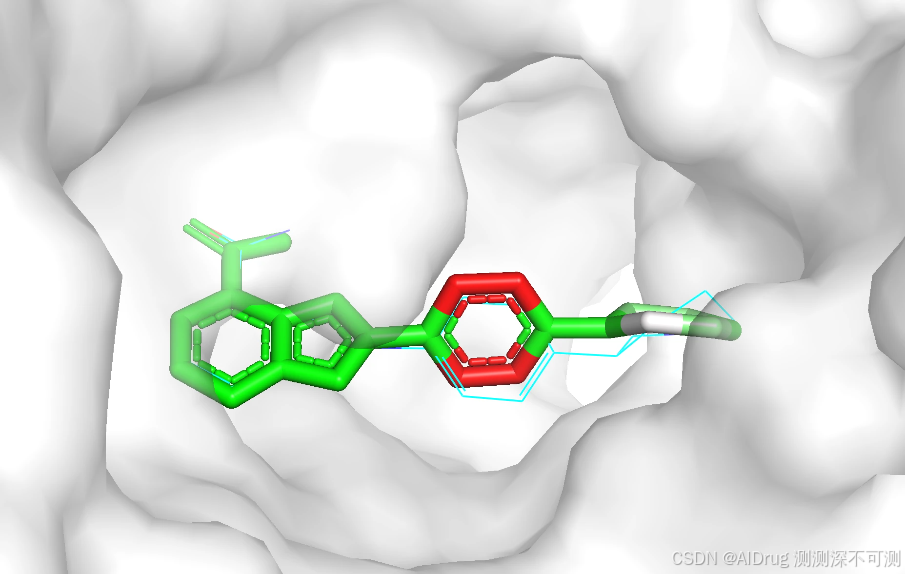
3 directories, 3 files随机数设置为 42 的预测对接构象为 ./4R6E/structure_output_42/0.pdb ,查看 ./4R6E/output_42.csv 表格得知该对接构象预测的结合亲和力为 -7.71 ,预测的对接模型置信度为 0.77。预测的对接构象如下所示,蓝色是参考配体,模型预测的对接构象通过颜色展示模型的置信度(红白绿颜色过渡表示 0-1)。两个构象的位置基本一致,证明在该测试案例上的整体预测效果良好,但该配体中间的环结构有一定的摆动。
随机数设置为 425 的预测对接构象为 ./4R6E/structure_output_425/0.pdb ,查看 ./4R6E/output_425.csv 表格得知该对接构象预测的结合亲和力为 -7.72 ,预测的对接模型置信度为 0.72。生成结构如下:
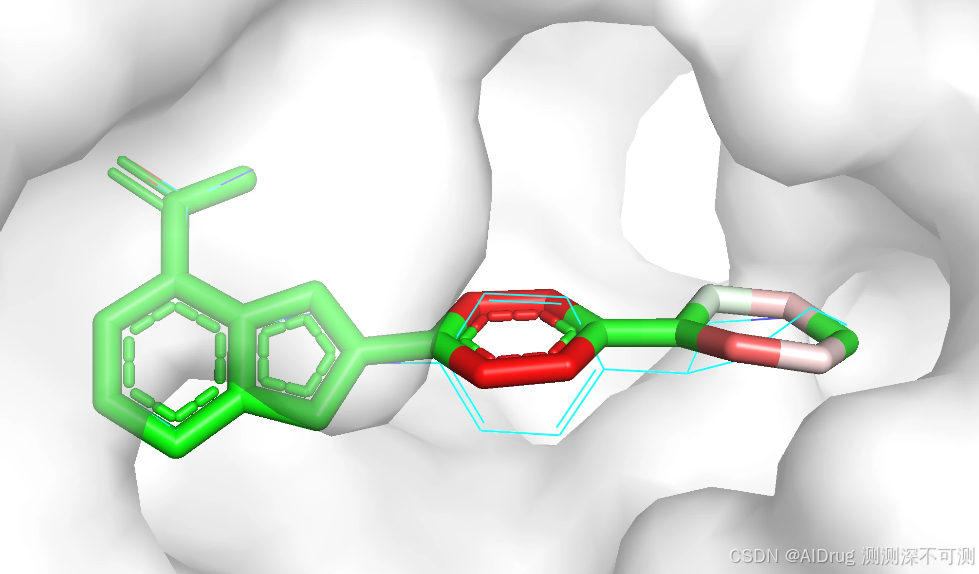
随机数设置为 425 的预测对接构象为 ./4R6E/structure_output_2024/0.pdb ,查看 ./4R6E/output_2024.csv 表格得知该对接构象预测的结合亲和力为 -7.72 ,预测的对接模型置信度为 0.79,结果如下:
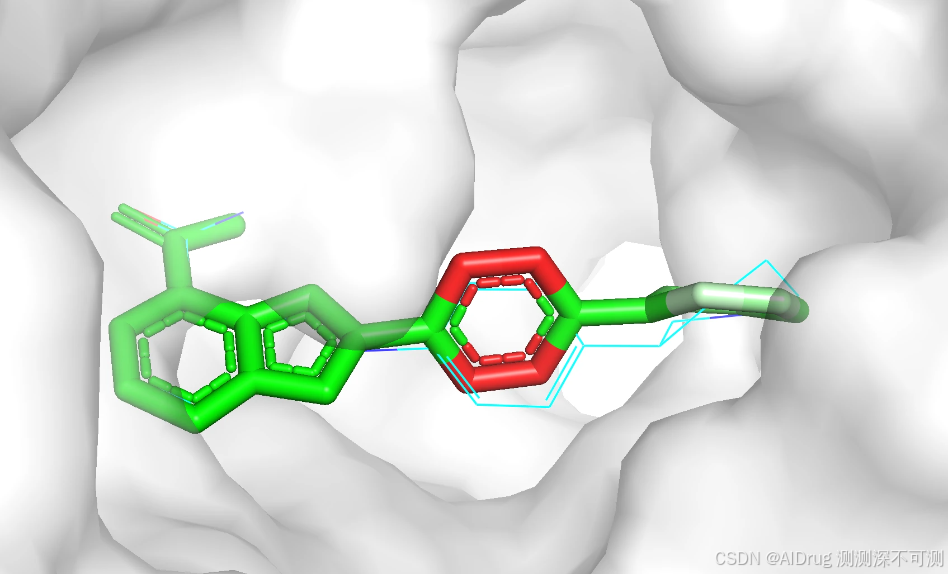
下图展示在蛋白口袋中的预测的三个构象和晶体结构(黄色),模型预测的对接构象通过颜色展示模型的置信度(红白绿颜色过渡表示 0-1)。可以看出,相对于晶体结构,预测的构象主要是中间和右端的环结构有一定摆动。左端的并环结构位于较为狭窄的亚口袋区域,可以摆动的自由度较小。中间和右端的结构所处的口袋空间比较开阔,允许摆动的自由度较大。小分子可以摆动的程度与模型输出的置信度趋势一致。三个不同的随机数产生的小分子构象存在一定的差异,但高置信度区域的结果基本一致,低置信度存在一定的差异。
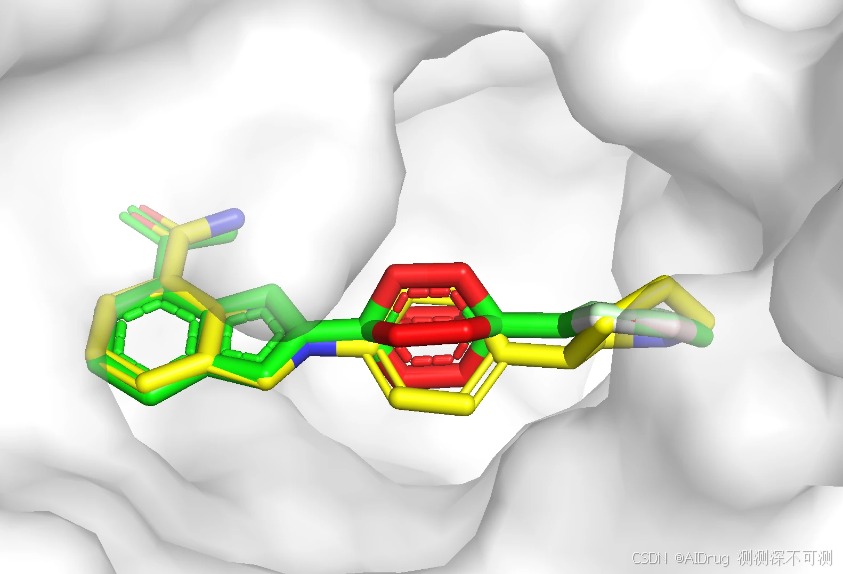
我们把预测的三个构象分别命名为 pose_pred_42.sdf、pose_pred_425.sdf 和 pose_pred_2024.sdf 并保存到 ./4R6E/pose_pred 文件夹中,原始配体构象保存为 ./4R6E/pose_pred.sdf 。创建 CalcLigRMSD.py ,定义 CalcLigRMSD 函数用来计算参考配体和预测的对接构象之间的 RMSD 。
预测的三个构象(seed=42,425,2024)和配体的晶体结构之间未对齐的 RMSD 都小于 1 Å,表明预测构象和晶体结构基本重合,模型预测的效果良好。
RMSD(seed=42): (0.51, 24)
RMSD(seed=425): (0.66, 24)
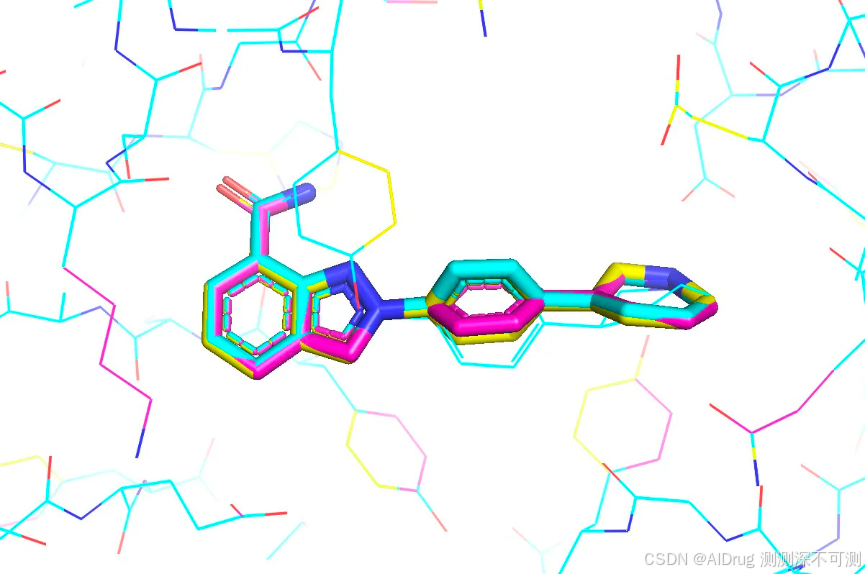
RMSD(seed=2024): (0.52, 24)我们设置三个不同的随机数发现小分子三个对接构象之间不完全相同,置信度较低的片段结构部分在口袋中有一定摆动。但三个不同的随机数对应的蛋白的主/侧链完全相同,如下图:
此外,模型可以使用分子 SMILES 作为输入,并通过 RDKit 生成分子构象。当使用 SMILES 作为分子输入时,首先会使用 AllChem.EmbedMolecule 来为该分子生成一个初始的三维构象。接着,使用 MMFF(Merck Molecular Force Field)对分子进行能量优化。优化时,原子的最大位移限制为 1 Å。最后,执行 20 次最大化步骤的能量最小化。至此,从分子 SMILES 得到分子构象。根据 RDKit 生成的构象再预测对接到蛋白口袋中的构象。
3.2.2 测试体系 ABL1 介绍
为了测试 FlexPose 的真实对接能力,我们设置了 ABL1 体系。其中,包含了两个蛋白(口袋结构),一个是 3KF4_A.pdb,属于 Apo 结构,其底物口袋和变构口袋的构象均不能直接结合小分子。另一个是 6HD6_allo_protein.pdb,属于 Holo 结构,在此结构中,存在两个小分子,一个底物小分子抑制剂 Imatinib,另一个是变构小分子 Compound 6。
3KF4_A.pdb Apo构象(紫色) 和 6HD6_allo_protein.pdb Holo 构象(Cyans)在底物口袋对比,有明显的 loop 的变化:

3KF4_A.pdb Apo构象(紫色) 和 6HD6_allo_protein.pdb Holo 构象(Cyans)在底物口袋对比,可以看到有明显的 α-helix 的偏折:
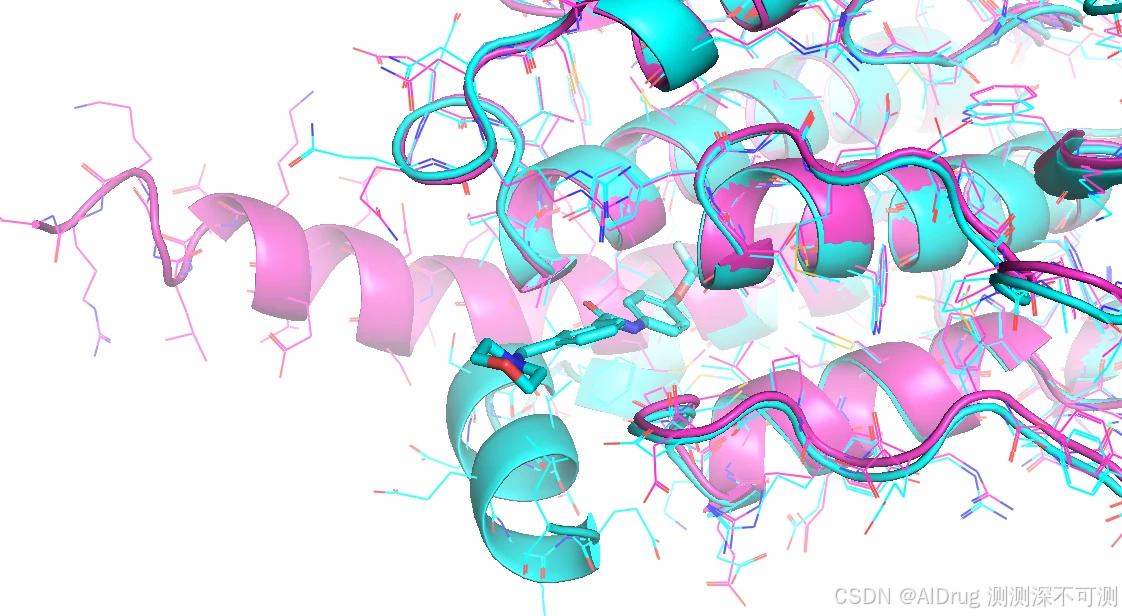
除了底物分子 Imatinib 和 变构分子 Compound 6,我们还找到了如下分子。其中, Compound 5 和 Compound 7 均属于变构分子,且活性与 Compound 6有明显的区别。Compound X 则是在变构分子的基础上,设计出来的,我们认为这个分子没有活性或者活性很低。Compound N 则是来源于非激酶体系的变构小分子,是肯定不能结合在底物或者变构位点的。
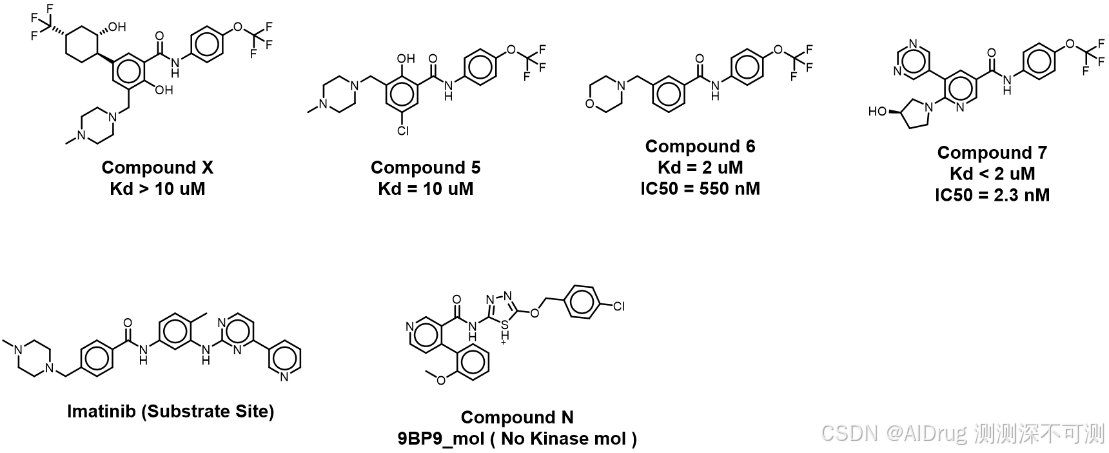
关于变构口袋的形状,在 Holo 结构下,小分子大部分在封闭口袋内,另一半在半溶剂区。
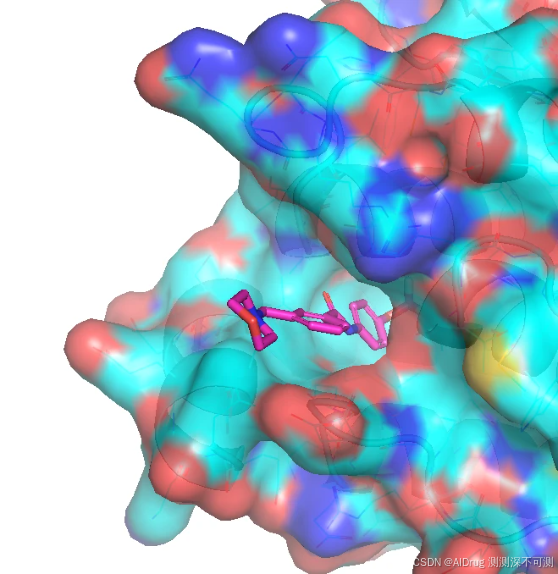
在同一视角的 Apo 结构下,口袋也有一定的形状,但是半溶剂区的面积更大,主要原因是 α-helix 没有出现弯折,口袋并没有很好成型。
接下来,我们尝试将所有的小分子都通过 FlexPose 对接到变构口袋中,分别对 Apo 进行测试。
3.2.3 ABL1 Apo 结构 (3KF4_A.pdb) 作为输入
首先,分别创建 ./ABL1/compound 和 ./ABL1/proteion 文件夹,./ABL1/compound 中存放要预测对接构象的 ABL1 中的 6 个分子, ./ABL1/proteion 中存放 3KF4_A.pdb 和 6HD6_allo_protein.pdb 。采用内置案例的方法,得到个对接以后的 pose。
模型预测结束后,预测结果保存在 ./ABL1/result_3KF4_cpd5 文件夹中,文件目录如下。每个文件夹分别对应一个分子在蛋白口袋中的构象,以及预测的结合亲和力结果。
.
|-- structure_prediction_3KF4_9BP9_mol
|-- structure_prediction_3KF4_Imatinib_2D
|-- structure_prediction_3KF4_compound_5
|-- structure_prediction_3KF4_compound_6
|-- structure_prediction_3KF4_compound_7
`-- structure_prediction_3KF4_compound_X
6 directories, 0 files对接构象和化合物 6 (晶体) 的 RMSD 的输出结果如下(括号内的元组分别对应 RMSD 和匹配的最大原子数目):
RMSD(pred_3KF4_cpd5.sdf): (0.74, 26)
RMSD(pred_3KF4_cpd6.sdf): (0.64, 27)
RMSD(pred_3KF4_cpd7.sdf): (1.22, 16)
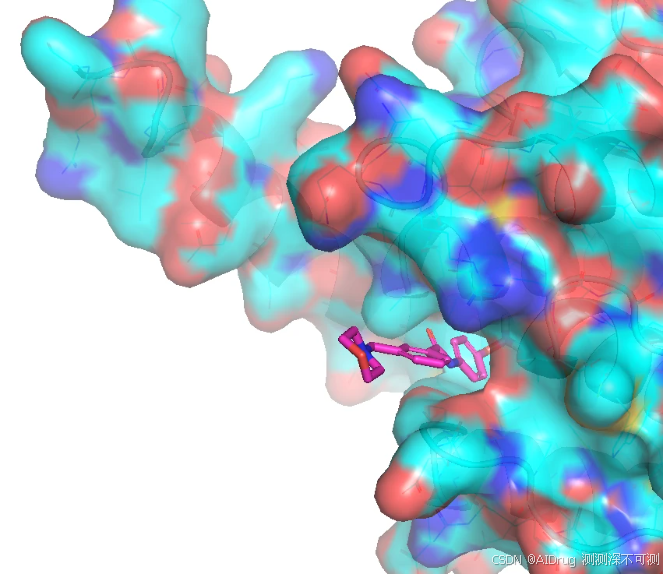
RMSD(pred_3KF4_cpdX.sdf): (0.68, 26)所有生成的小分子位置和朝向都比较一致,这显然是因为 FlexPose 指定了口袋以及口袋形状比较狭窄的缘故。如下图:
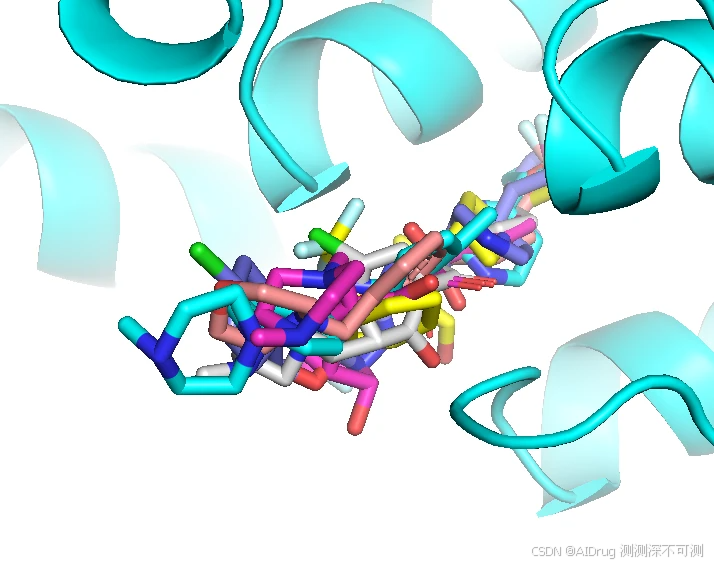
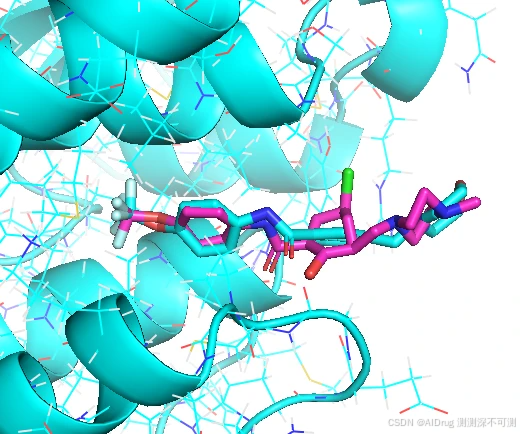
其中,FlexPose 生成的 Compound 5 (紫色) 和 Compound 6 (蓝色)的结合 pose 很相近,与晶体的 compound 6 的 pose (黄色)也非常接近,RMSD 为 0.74 和 0.64。特别是三氟甲基的末端(左侧)。但是由于苯环上的 Cl 取代和 OH 取代导致右侧的 pose 有所差异,苯环不在一个平面上,如下图。
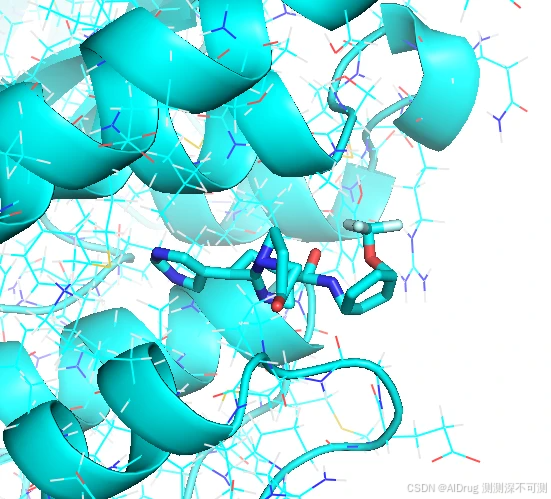
但是 Compound 7 则出现了奇怪的翻转,同时小分子的构象非常扭曲,如下图。
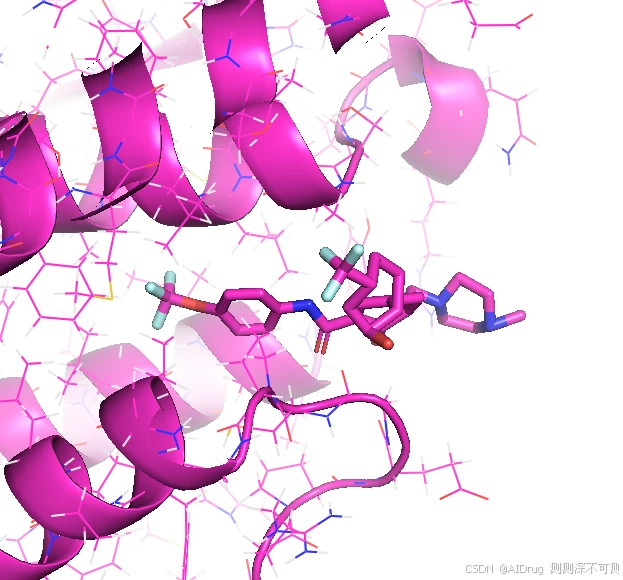
活性更差的 Compound X 的结果和 Compound 7 类似,小分子也是非常的扭曲,如下图:
对于有活性的 Compound 5, Compound 6, Compound 7, Compound X FlexPose 输出的小分子置信度着色依次如下。
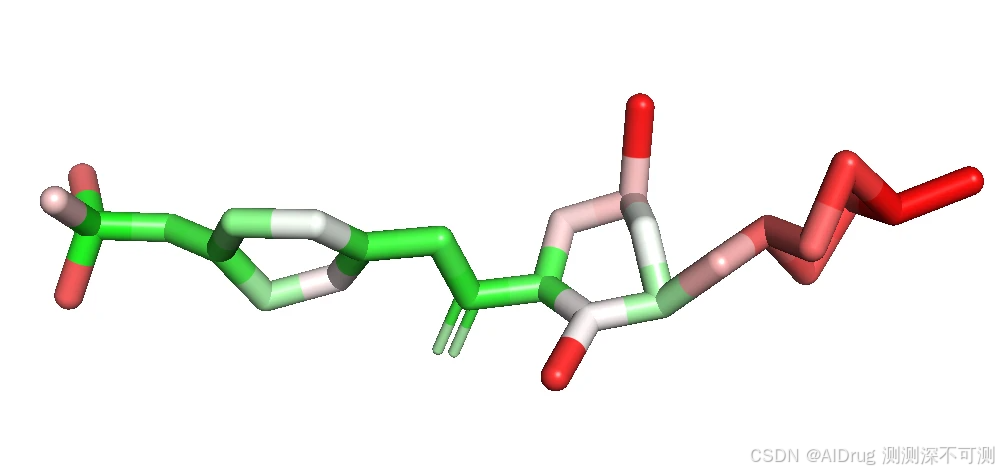
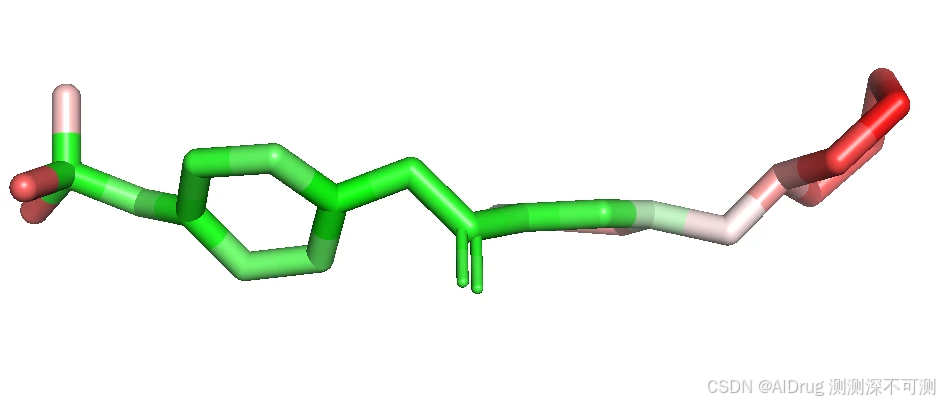
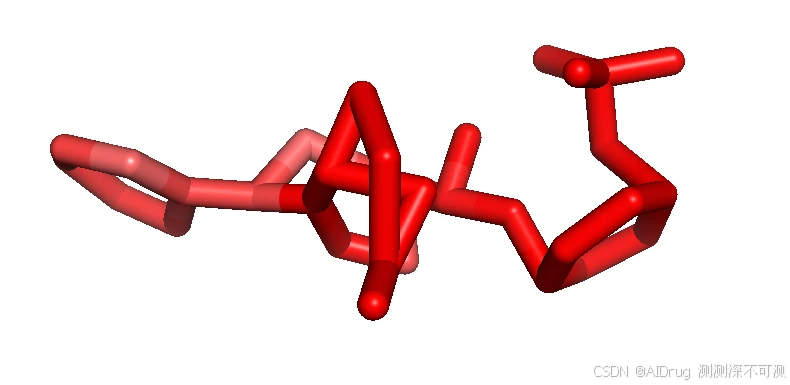
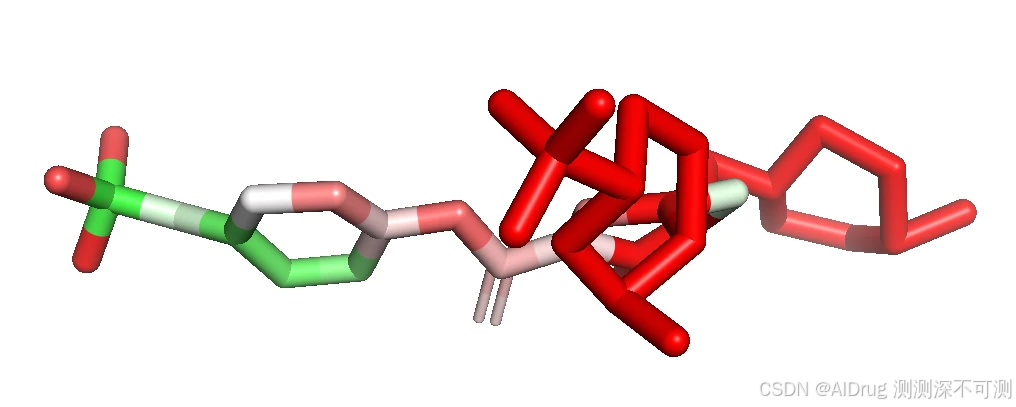
而对于两个不活的分子 Compound N 和 Imatinib ,也可以被 FlexPose 插入到口袋中,输出一些可能的构象。分别依次如下如图:
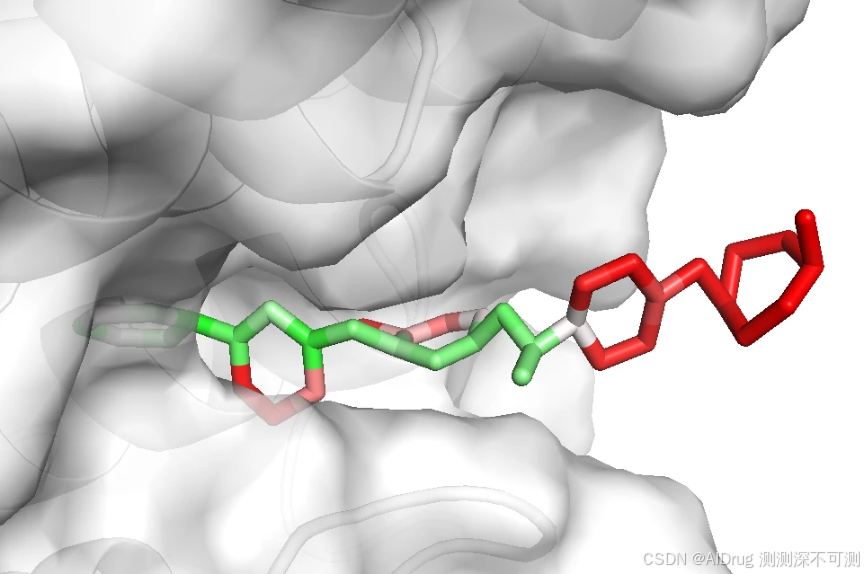
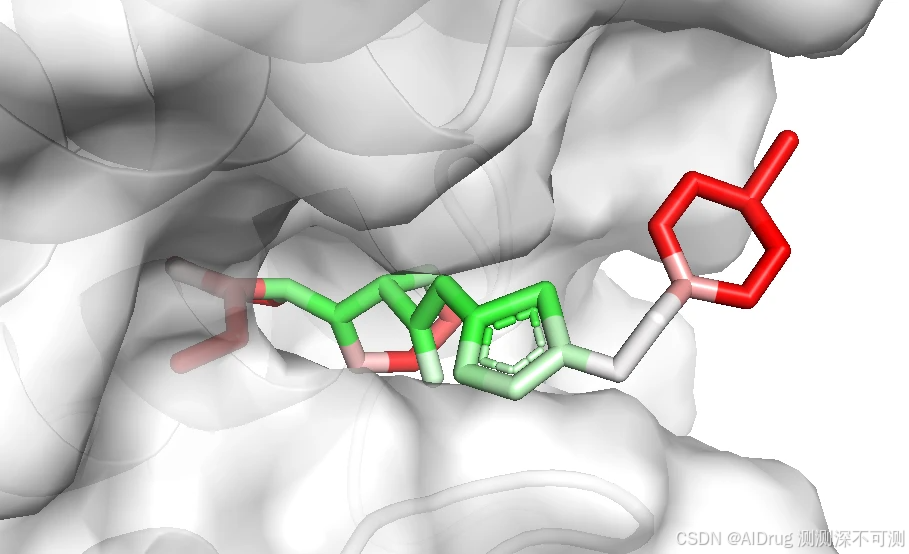
从上述结果来看,置信度模型对于溶剂区都是识别为高不可信,而封闭口袋则是根据相互作用来判断,这说明,FlexPose 的置信度模型确实可以帮助判断生成 Pose 的合理性。
以下是 FlexPose 在变构口袋上预测的各个小分子预测的置信度、预测的亲和力,以及小分子原来的Kd、IC50, 见下表:
| IC50 (uM) | Kd (uM) | 预测置信度 | 预测亲和力 | RMSD | |
| Compound 5 | / | 10 | 0.46 | -5.27 | 0.74 |
| Compound 6 | 550 | 2 | 0.56 | -4.05 | 0.64 |
| Compound 7 | 0.0023 | <2 | 0.04 | -6.14 | 1.22 |
| Compound X | / | >10 | 0.19 | -7.6 | 0.68 |
| Compound N | False | False | 0.38 | -5.6 | / |
| Imatinib | False | False | 0.37 | -6.04 | / |
注:False 为不活没有活性,RMSD 为,与晶体中 Compound 6 的最大公共子结构的 RMSD,即参考分子为晶体中的 compound_6.sdf。
另外,我们注意到 FlexPose 输出的蛋白口袋的构象,相较初始的输入构象,并没有区别。虽然文章中指出,此模型针对 apo 到 holo 结构的 CA 原子位置平均误差低至 0.78,但我们在这个体系上没有看到明显的口袋构象的变化,基本完全一致,除了个别侧链有很小很小的区别。如下图,紫色为初始构象,和 FlexPose 的输出构象(蓝色)几乎完全相同,说明,FlexPose 柔性的对接柔性程度很低,蛋白口袋几乎刚性的。

PoseBuster 的评估结果来看,这六个分子和蛋白之间的最小距离较近,因为指定的蛋白口袋是一个比较狭窄的空间。化合物 5 和 7、imatinib 的对接结构没有通过键角的检查。化合物 5 和 7 的对接结构的分子内能量检查也没有通过,化合物 7 的对接结构中非共价键结合原子对之间的原子间距离过近,和蛋白存在空间冲突的问题。
此外,我们还对 FlexPose 在 ABL1 holo 结构上的性能也做了测试,详情参考测评文档。PoseButser 以及 对接后分子 RMSD 的计算方法,也请参考测评文档文档。
四、总结
FlexPose 是一个高效的端到端深度学习框架,用于蛋白质-配体复合结构的柔性建模(柔性对接)。FlexPose 模型的特点是,标量-向量双特征表示、SE(3) 等变网络、构象感知预训练和弱监督学习。结果上,FlexPose 的准确性显著超越了所有测试过的对接工具和混合深度学习方法,且大大减少了所需时间。此外,FlexPose 还提供准确的结合亲和力和模型置信度的估计,能够辅助后续分析,对药物相关的应用(如药物设计和筛选)非常有帮助。
与大多数现有方法相比,FlexPose 能够在不依赖传统对接策略(采样和评分)的情况下对复杂结构进行柔性建模。对于排名第二和第三的基于深度学习的方法(EDM-dock 和 RTMscore),它们仅基于标量特征预测蛋白质残基与配体原子之间的成对空间关系(距离矩阵或似然势)。这种设计更隐性地考虑了柔性,使得其几何表示相对于 FlexPose 更简化。
此外,FlexPose 提供了两个功能:结合亲和力和模型置信度,有助于量化结构-性质/活性关系(QSPR/QSAR)的机制识别。
完整测评文档以及代码修改,详见:https://download.csdn.net/download/wufeil7/90043910?spm=1001.2014.3001.5503



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











