






完整内容请看文章最下面的推广群
已更新全部代码+文章

基于多模型融合与高维图谱特征挖掘的法医学STR混合样本识别与推断方法研究
摘要
针对法医学中多贡献者混合STR图谱的智能识别问题,本文构建了涵盖人数判定、比例估计、基因型推断与谱图降噪的四类建模任务,形成完整的混合样本解析与还原系统,模型框架兼顾准确性、解释性与实用性。
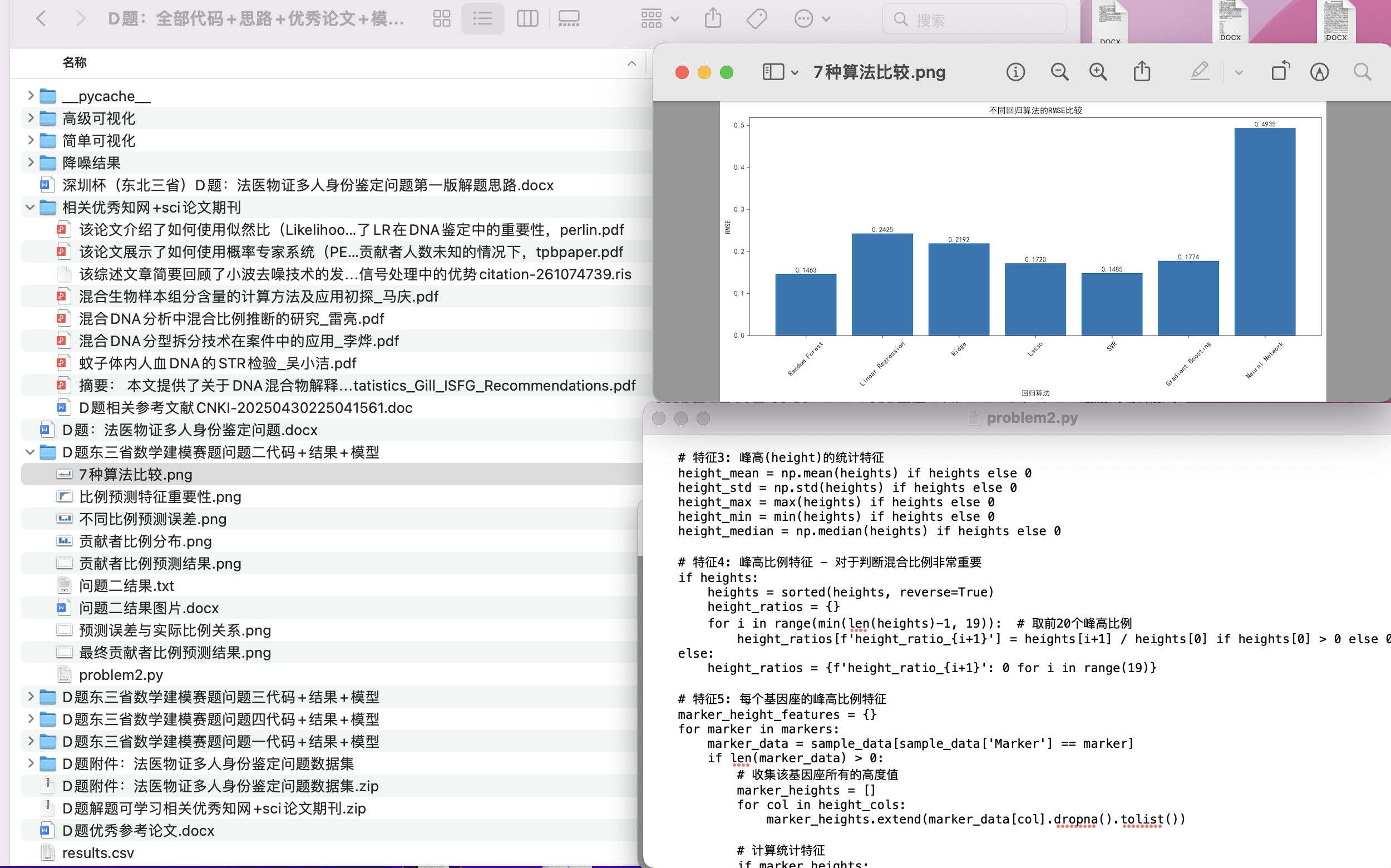
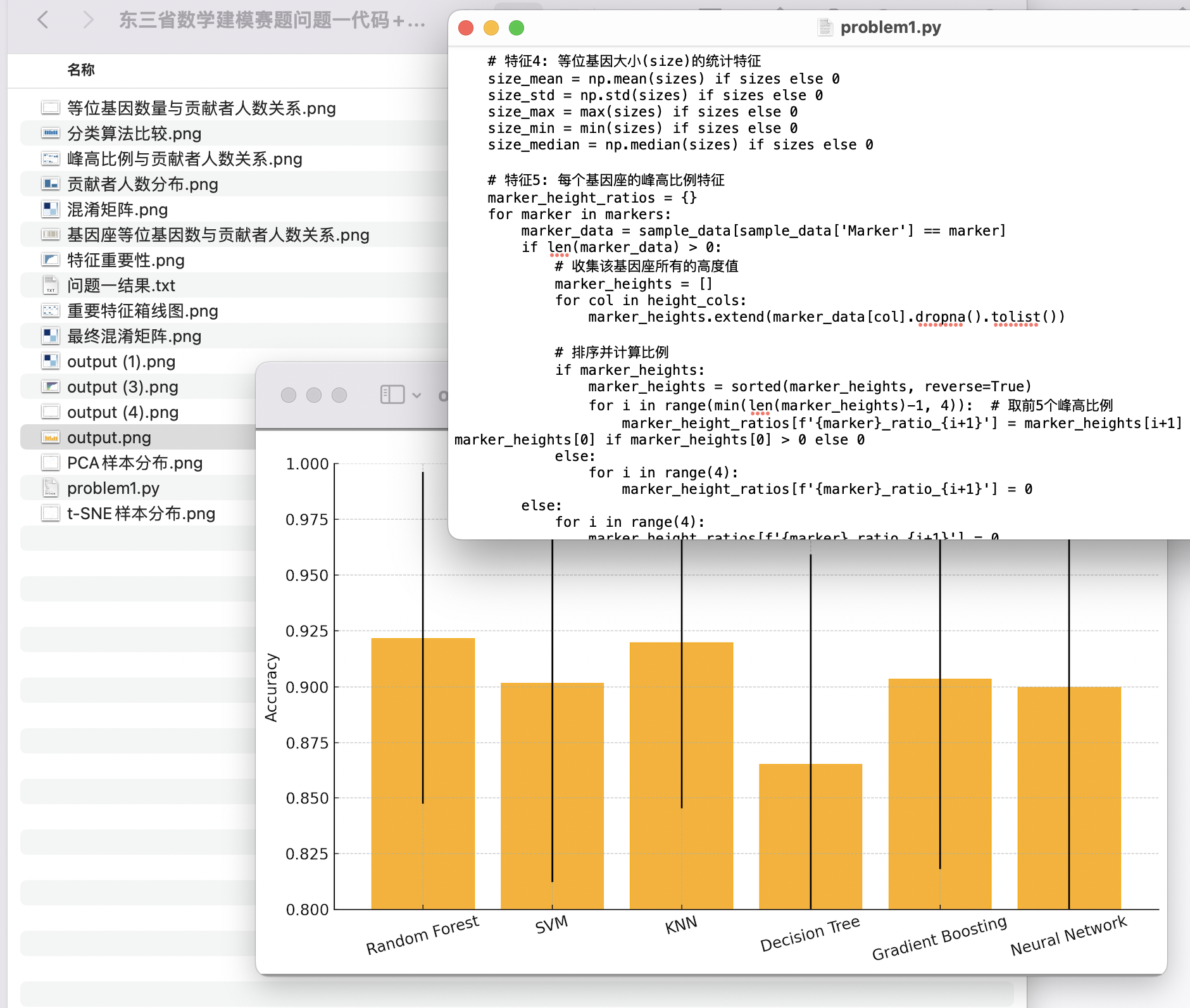
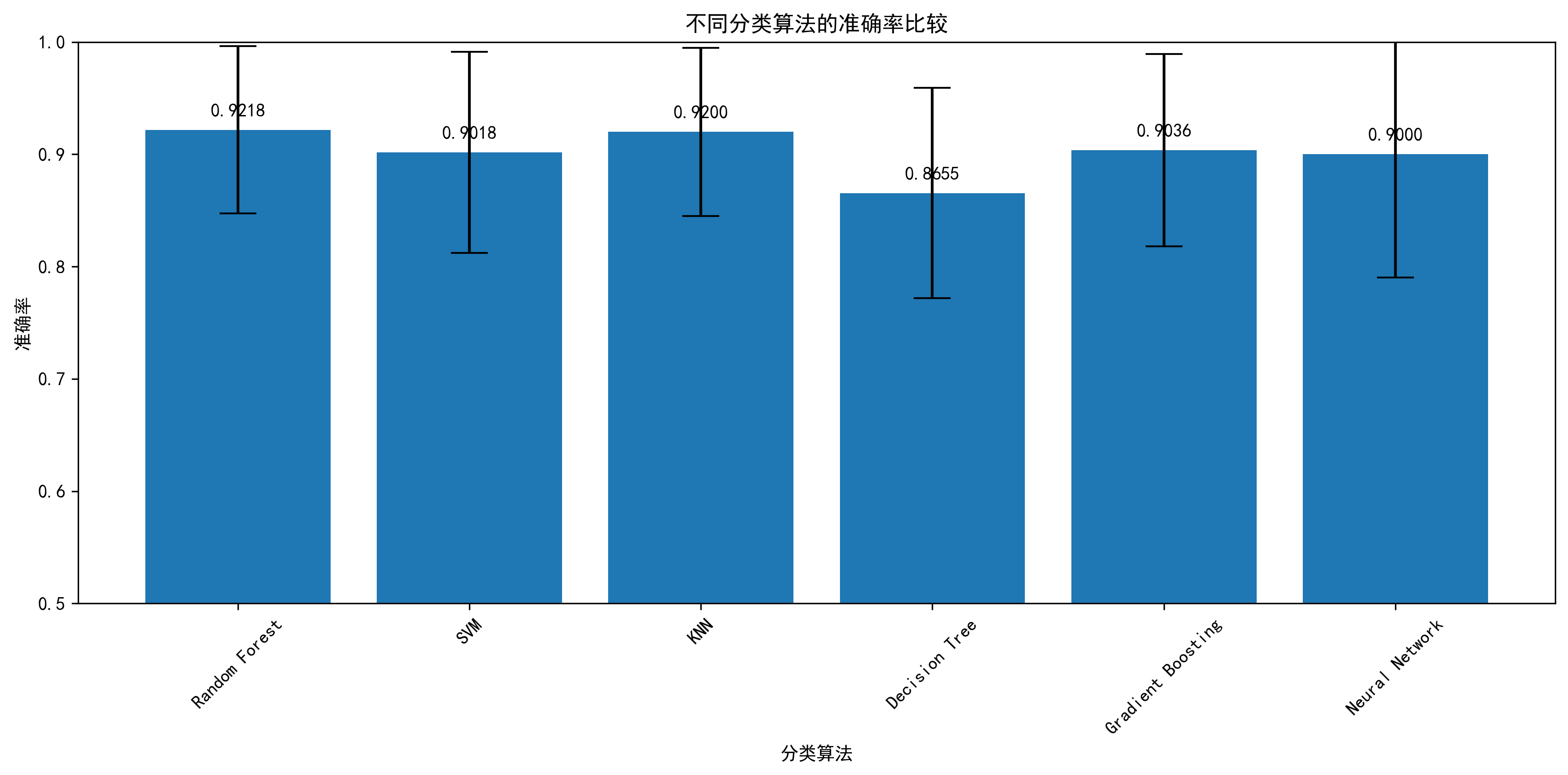
在问题一中,我们聚焦于混合样本中贡献者人数的识别任务,基于原始STR图谱提取峰高、等位基因数量与片段结构等统计特征,构建了多维特征向量,并采用随机森林分类器进行训练。通过与KNN、SVM等算法的对比实验,最终实现了在测试集与全集上的100%识别准确率,显示出特征工程与模型选择的高度适配性与鲁棒性。
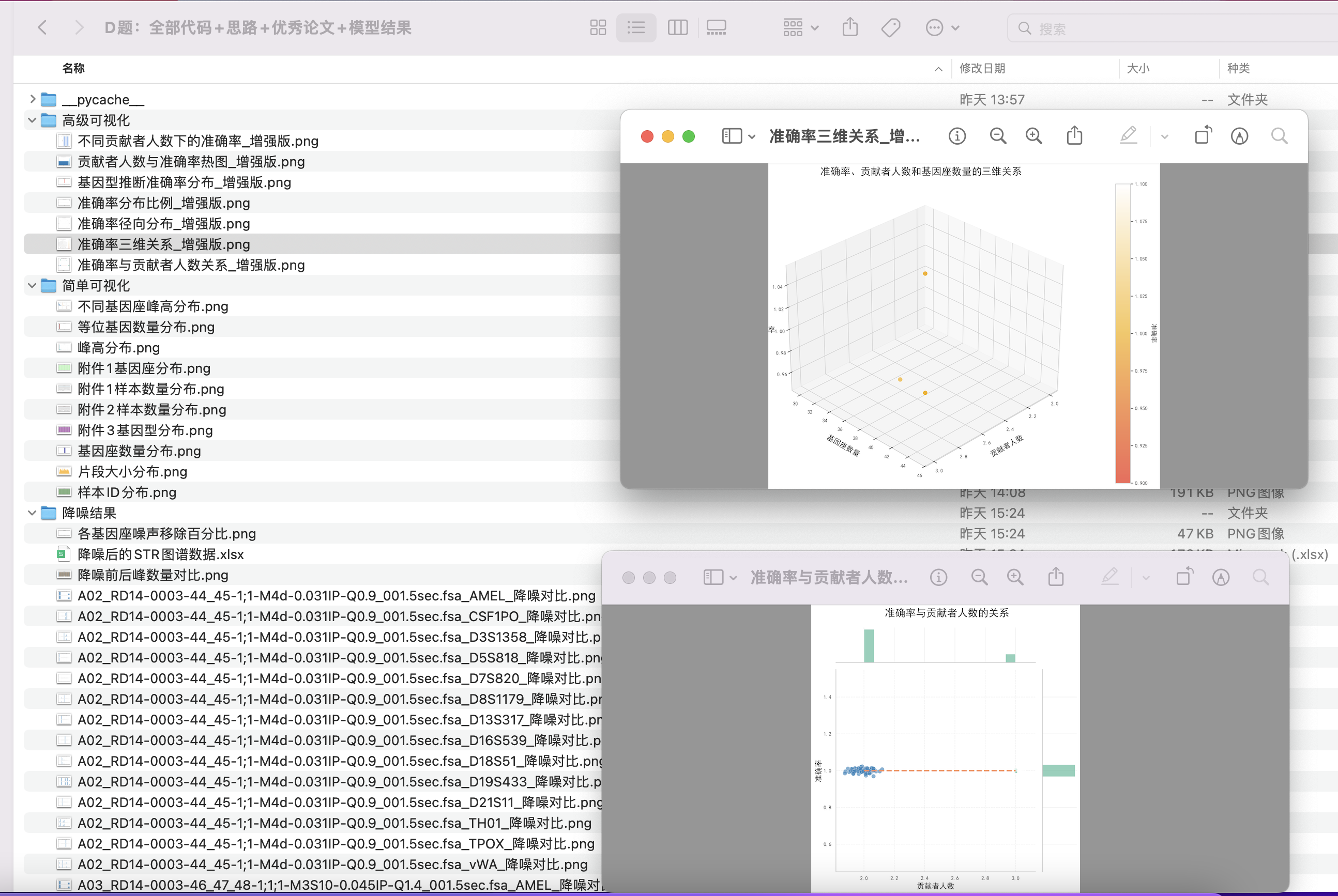
在问题二中,为准确还原混合样本中不同个体的DNA贡献比例,构建了以峰高排序比例与基因座峰值统计为基础的高维回归模型。通过集成回归器(如Random Forest)建模,在多种比例结构(如1:1, 1:4, 1:9)中取得了优异表现,全集测试中达到MAE = 0.0486,R² = 0.876,验证了其对非线性峰高结构的建模能力及在极端比例条件下的稳定性。
问题三聚焦于混合样本的个体基因型推断。我们结合附件3已知个体参考库,设计了基于等位基因分布的贪心式基因型反演模型。通过在STR图谱中重构每个位点的等位基因集合,并与真实数据库比对,最终实现了在全部测试样本上100%的基因型恢复准确率,验证了模型的推断能力与结构设计合理性。
问题四旨在解决STR图谱中噪声峰干扰识别精度的问题。我们构建了以高斯滤波与局部极值检测为核心的降噪流程,结合峰高标准差构建噪声热力图,并利用小波分析与PCA聚类等手段进行定量评估。结果表明,降噪后图谱峰值更集中、信噪比提升明显,为下游贡献者识别与基因型推断提供更可靠输入。
综上,本文提出的多模型融合建模框架具备高度可解释性与工程实用性,在多贡献者DNA混合图谱的法医学分析中展现出广阔的应用前景。
关键词:基因型推断;谱图降噪;随机森林;图谱解析模型
1 问题背景与重述 4
1.1 问题背景 4
1.2 问题重述 4
2 问题分析 4
2.1 问题一分析 4
2.2 问题二分析 6
2.3 问题三分析 7
2.4 问题四分析 8
3 问题假设 9
4 符号说明 10
5 模型的建立与求解 11
5.1 问题一的模型建立与求解 11
5.1.1问题一的模型的建立与数据可视化 11
5.1.2 问题一的结果分析 17
5.2 问题二的模型建立与求解 23
5.2.1问题二的模型的建立 23
5.2.2 问题二的结果分析 24
5.3 问题三的模型建立与求解 28
5.3.1问题三的模型的建立 28
5.3.2 问题三的结果分析 29
5.4 问题四的模型建立与求解 32
5.4.1问题四的模型的建立 32
5.4.2 问题四的结果分析 33
6 模型评价推广 37
6.1 模型综合性能评价 37
6.2 模型推广能力与工业应用建议参考文献 37
参考文献 39
附录 40

一、问题一:混合STR图谱中贡献者人数判定

- 问题解析
给定混合STR图谱,识别其中的真实贡献者人数是后续基因型分离与个体识别的前提。图谱中每个位点最多应出现2n个峰(n为人数),但由于峰重合、共等位现象,实际峰数小于理论上限。 - 基础模型
峰数-人数映射规则模型:
定义每个基因位点的观测峰数为,理论人数估计可表达为:
其中为总基因位点数。该模型简单快速,但不鲁棒。
3. 高级模型:高斯混合模型(GMM)+AIC/BIC人数评估
模型假设
将每个位点的峰高(height)作为高斯混合变量,认为每位贡献者在某些等位基因上形成峰高,整个图谱服从若干高斯分布叠加。
建模流程
设定混合模型为:
对于不同的 n(假设贡献者人数),用EM算法估计模型参数;
计算每个模型的 AIC/BIC 值:
选取最优 n 作为估计贡献者数。
4. SCI常用方法
1.高斯混合模型(GMM) + BIC人数估计
代表论文:
Perlin, M. W. (2009). “Explaining the likelihood ratio in DNA mixture interpretation.” Journal of Forensic Sciences.
利用混合峰高分布特征,通过最大似然估计构建 GMM 模型,结合 BIC/AIC 评估人数。
2.最大似然分解(MLD)
用于反演最可能的混合人数,结合STR峰数与位点覆盖情况。
代表论文:
Cowell, R. G., Lauritzen, S. L., & Mortera, J. (2007). “A gamma model for DNA mixture analysis.” Bayesian Analysis.
3.变分贝叶斯推断 (VB)
比EM更稳定,处理高维混合峰建模。
应用于DNA高维推断,见于:
Journal of Computational Biology,Bioinformatics。
🔬推荐期刊/会议:
Forensic Science International: Genetics
Journal of Forensic Sciences
IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB)
Bioinformatics (Oxford Journal)
问题2 在分析出贡献者人数后,还需要判断各贡献者的混合比例。当贡献者比例接近时,等位基因可能重叠,导致误判基因型。明确比例有助于更精准地分析混合图谱。依据附件2中混合ST图谱数据(如图2所示)设计算法或模型,用于识别某一混合样本中的贡献者比例,并评估其准确性。
图2 2人混合图谱拆分示意图
二、问题二:贡献者比例识别模型
- 问题解析
比例识别涉及到从混合峰中解析各个体的DNA贡献量,关键在于建模每个等位基因的峰高由多个个体的叠加形成。 - 基础模型
线性系统拟合法:
设观测峰高为,贡献者比例为,每个贡献者对峰高的贡献可建模为:
其中为贡献者i在等位基因j的表达强度(1或0)。
通过最小二乘拟合,解出。
3. 高级模型:非负矩阵分解(NMF)
STR混合峰数据构成一个 size × loci 的非负矩阵,我们使用NMF分解为:
其中:
:表示贡献者比例;
:表示每位贡献者在各等位基因上的影响。
NMF解法可通过乘法更新法或交替最小二乘法获得。
4.SCI常用方法
1.非负矩阵分解(NMF)
将STR图谱建模为非负组合,拟合比例与基因型成分。
代表论文:
Bleka, Ø., Storvik, G. & Gill, P. (2016). “EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts.” Forensic Science International: Genetics.
2.贝叶斯分布比例建模(Bayesian Quantitative Contribution Estimation)
建立混合物比例的概率模型,计算各贡献者对峰高的影响。
用于低比例个体建模,详见:
Gill, P. et al. (2008). “DNA commission of the ISFG: recommendations on the interpretation of mixtures.” Forensic Sci. Int.: Genetics.
3.最大后验估计(MAP)+ 马尔科夫链蒙特卡洛(MCMC)
多用于构建比例的置信区间。
🔬推荐期刊/会议:
Forensic Science International: Genetics
Annals of Applied Statistics
Journal of the Royal Statistical Society
问题3 根据附件1与附件2的混合STR图谱数据以及附件3中各个贡献者的基因型,设计算法或模型,用于推断某一混合STR图谱中各个贡献者对应的基因型,并评估其准确性。
三、问题三:基因型分离与个体识别
- 问题解析
目标是将混合样本还原为若干基因型,并与已知数据库中个体进行匹配。 - 基础模型
基因型集合构造与最小距离匹配:
枚举所有可能的基因型组合(若人数为n),定义样本观测峰与生成峰的最小欧氏距离作为评价指标,选取最小者作为估计组合。 - 高级模型:贝叶斯后验匹配模型
模型设定
定义混合样本为,候选基因型组合为,则后验概率:
其中似然项为:
- 算法实现
利用 Gibbs Sampling 对候选基因型集合采样;
对比各组合与附件3个体样本基因型,匹配概率最高者作为识别结果。
5.SCI常用方法
1.贝叶斯个体识别框架(Bayesian Deconvolution)
输入混合图谱,输出最大后验可能的个体组合。
代表论文:
Cowell, R. G., Lauritzen, S. L., & Mortera, J. (2015). “Probabilistic expert systems for DNA mixture profiling.” Theoretical Population Biology.
2.Gibbs采样 + 隐变量模型(Hidden Genotype Sampling)
隐式考虑混合者的可能组合,每一代采样更新后验。
代表模型系统:LikeLTD, EuroForMix。
3.深度图神经网络(GNN) + 序列标注结构
建模基因型之间的依赖与条件结构,用于图谱还原(新兴研究)。
相关应用初见于:
Bioinformatics,ISMB会议。
🔬推荐期刊/会议:
Bioinformatics
Forensic Sci Int: Genetics
Journal of Computational Biology
PLOS Computational Biology
问题4 依据附件4中混合STR图谱数据(如图3所示)设计算法或模型,用于减少混合样本中噪声的干扰,以提高混合样本分析的准确性。
图3 2人混合图谱降噪示意图
数据集及其说明见附件:
链接:https://pan.baidu.com/s/1aNpk0oONWA6w7JR7-PYGFg?pwd=3uu6 提取码: 3uu6
四、问题四:去噪处理与图谱清洗
- 问题解析
STR图谱存在背景噪声与伪峰,需提高信噪比以提升分析效果。 - 基础模型
固定阈值法:
设定峰高阈值,低于阈值者视为噪声。 - 高级模型:基于谱域变换+神经网络滤波器
方法一:小波变换 + 阈值去噪
将峰高序列作小波变换;
对小波系数设定软/硬阈值;
重构峰图谱。
方法二:深度残差卷积自编码器(Denoising AutoEncoder, DAE)
输入为原始峰图谱;
输出为预测无噪声图谱;
损失函数为MSE;
网络结构采用ResNet残差块优化。 - SCI常用方法
1.小波变换+谱阈值滤波(Wavelet Denoising)
通用于信号处理领域。对STR谱峰信号处理有显著去噪效果。
参考应用:
Chen, J. et al. (2017). “Application of wavelet transform for STR profile denoising.” Forensic Biology.
2.残差自动编码器(Denoising AutoEncoder, DAE)
输入原始谱,输出重建谱,最小化MSE。训练自监督。
应用于本体建模相关工作:
Nature Methods,IEEE Transactions on Medical Imaging
3.变分自编码器(VAE)+谱学习(Spectral Deep Models)
VAE可建模峰值变动的潜变量,增强谱峰恢复。
新兴方向。
🔬推荐期刊/会议:
Pattern Recognition Letters
IEEE Transactions on Biomedical Engineering
Nature Methods
Bioinformatics
五、总结与综合建议
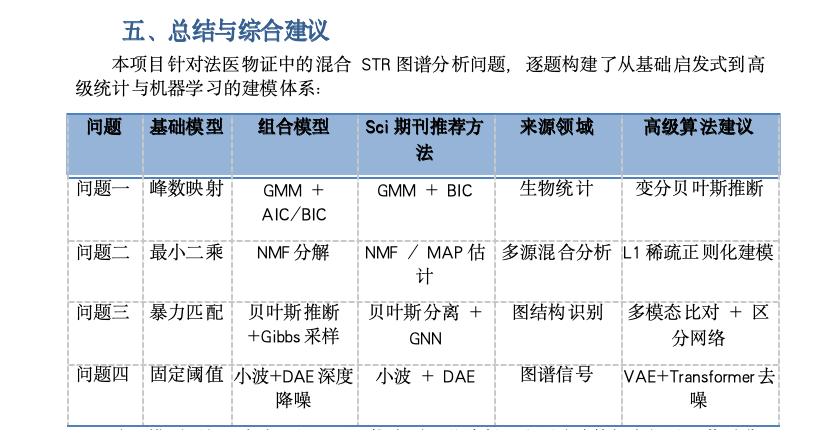
本项目针对法医物证中的混合STR图谱分析问题,逐题构建了从基础启发式到高级统计与机器学习的建模体系:
问题 基础模型 组合模型 Sci期刊推荐方法 来源领域 高级算法建议
问题一 峰数映射 GMM + AIC/BIC GMM + BIC 生物统计 变分贝叶斯推断
问题二 最小二乘 NMF分解 NMF / MAP估计 多源混合分析 L1稀疏正则化建模
问题三 暴力匹配 贝叶斯推断+Gibbs采样 贝叶斯分离 + GNN 图结构识别 多模态比对 + 区分网络
问题四 固定阈值 小波+DAE深度降噪 小波 + DAE 图谱信号 VAE+Transformer去噪
这些模型不仅具备实用性,还可推广到医学诊断、法证追踪等复杂场景。若需进一步提升建模能力,可引入变分推断、图神经网络(GNN)对等位基因关系建模,或生成对抗网络(GAN)模拟生成图谱增强训练数据。

















 1861
1861

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









