






KEGG KO的分析想必大家都不陌生,但是不知道什么原因,KEGG官方的各种文件都搞得遮遮掩掩,无论是做基因注释还是数据可视化,大家都非常难受。其中对于注释出来的KO号怎么进行KO号名称和代谢通路的注释,由于官方没有提供相应的一一对应表,所以对于数据分析与可视化来说十分不方便。。。。
好在KEGG官方并不是完全不给机会,提供了所谓的htext文件和网页让你进行分析,htext格式如下
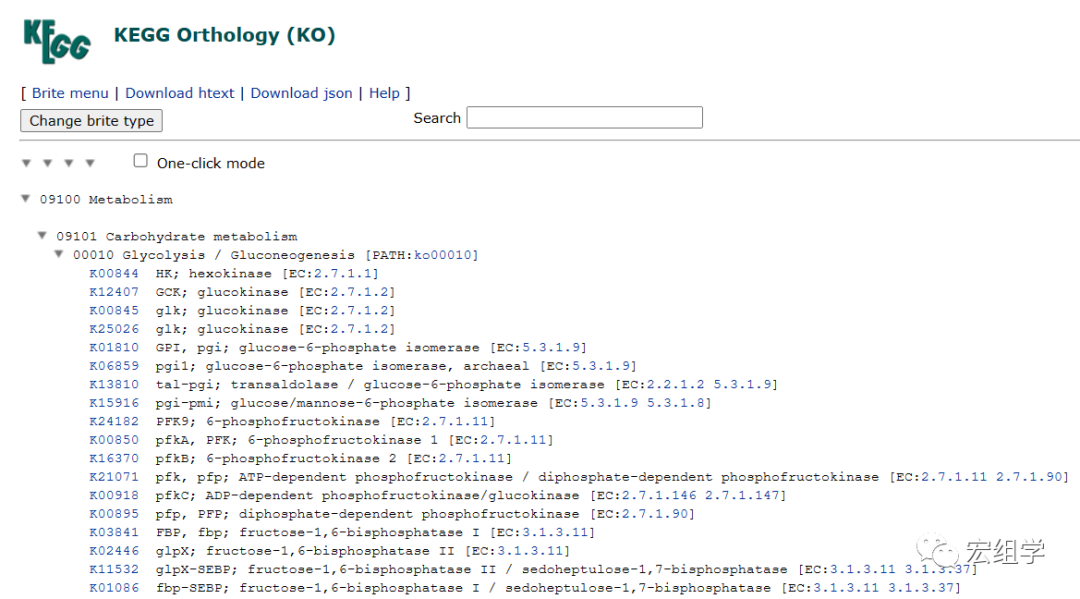
是不是看了感觉摸不到头脑?这玩意要怎么用来数据分析?
于是小编上网搜各种分析流程和教程之类的,也算是找到几个python或者perl脚本之类的,但是使用过后,发现要么就是出错,要么分析出来的效果不尽人意,所以小编打算自己动手写代码进行分析。因为小编目前对R比较熟悉,所以尝试着用R进行分析。废话不多说,具体代码和流程如下。
1. 下载htext文件
进入www.kegg.jp/kegg-bin/get_htext?htext=ko00001网站,点击左上角的Download json,下载json文件。
2. 开始使用R进行分析
1. 设置工作目录并读取json文件
setwd("~/代码驿站/自写代码/data")
library(rjson)
jsonData = fromJSON(file ="ko00001.json")
a=jsonData$name
b=jsonData$children读取得到的为list格式,如下、
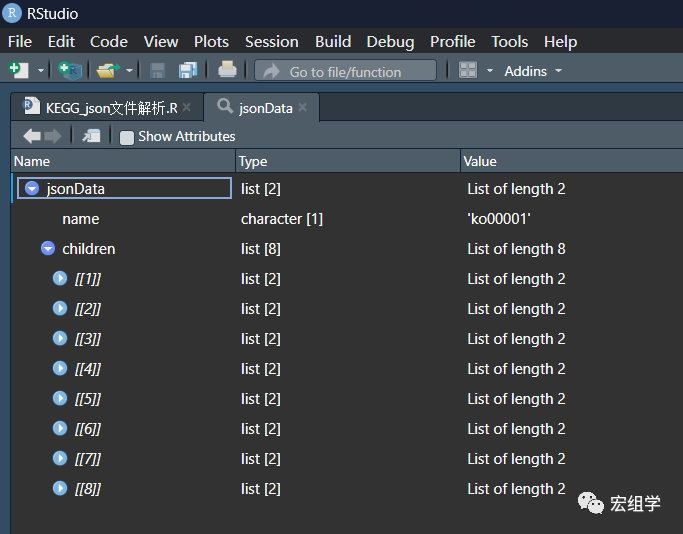
2. 解析list
list格式也算是R里面最复杂的格式,不过也没那么复杂,只要仔细观察,你就可以找到这个list里面的一些规律,有兴趣的朋友可以自己点进去看看,这里我不多说了,直接上代码。。
c=NULL
for (i in 1:length(b)) {
for (j in 1:length(b[[i]]$children)) {
for (w in 1:length(b[[i]]$children[[j]]$children)) {
for (x in 1:length(b[[i]]$children[[j]]$children[[w]]$children)) {
c=rbind(c,c(
b[[i]]$name,
b[[i]]$children[[j]]$name,
b[[i]]$children[[j]]$children[[w]]$name,
b[[i]]$children[[j]]$children[[w]]$children[[x]]$name
))
}
}
}
}这段代码就是解析json文件的核心代码了,接下来的工作大家也可以使用自己的需求进行写,小编在这里演示一下自己的方法。
3. 后期整理
library(stringr)
c=as.data.frame(c)
c$V1=substring(c$V1,7,nchar(c$V1))
c$V2=substring(c$V2,7,nchar(c$V2))
c$V3=substring(c$V3,7,nchar(c$V3))
#去除前6个用不到的字符(如果需要使用,则使用下面的代码先替换第一个空格,再进行分离)
library(tidyr)
d = separate(data=c, col = V3, into=c('V3', 'pathway_id'), sep ="[[]" )
d$pathway_id =str_replace_all(d$pathway_id,c("]"=""))
#分离出代谢通路ko号
d$V4 =sub(" ", "$", d$V4)
d = separate(data=d, col = V4, into=c('V4', 'KO_name'), sep ="[$]" )
#分离出K号
d$KO_name =sub(";", "$", d$KO_name)
d = separate(data=d, col = KO_name, into=c('gene', 'KO_name'), sep ="[$]" )
#分离出kegg的基因
d$KO_name =sub("[[]EC:", "$", d$KO_name)
d = separate(data=d, col = KO_name, into=c('KO_name', 'EC'), sep ="[$]" )
d$EC =gsub("[]]", "", d$EC)
#分离出每个K号的名称以及对应的EC编号这里为了避免出现错误的分离,因此小编首先进行了一些替换,然后再进行分离。
4. 出表!
names(d)[c(1,2,3,5)]=c("level1","level2","level3","KO")
write.table(d, file = "kegg pathway htext.txt", quote = F, sep = "\t", row.names = F, col.names = T)最后得到的表如下:
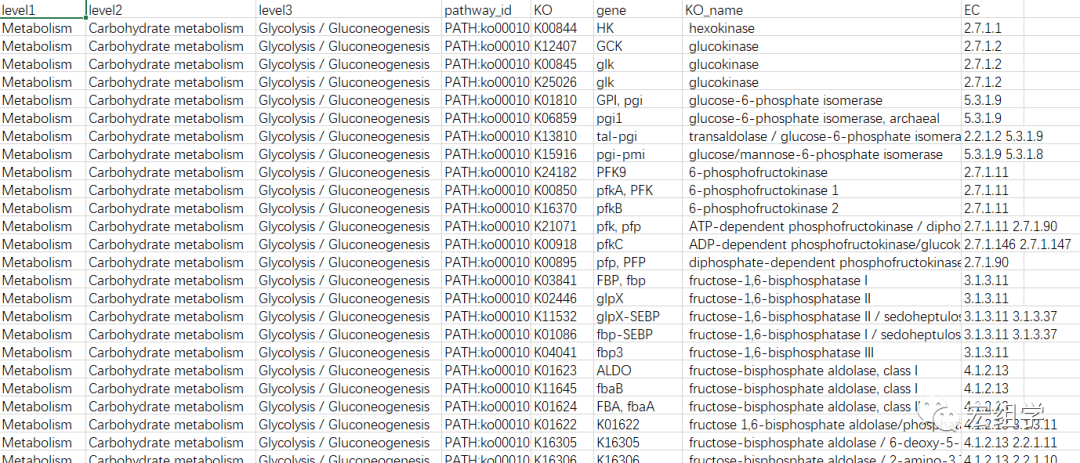
完美符合数据分析所需要的表格格式!
当然也有小伙伴可能会嫌麻烦,因此小编在这里也提供了一个脚本以及解析到的文件,地址为
更多内容,请关注公众号plantmicrobiome(宏组学)















 1232
1232











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









