







FASTA文件主要用于存储生物的序列文件,例如基因组,基因的核酸序列以及氨基酸等,是最常见的生物序列格式,一般以扩展名fa,fasta,fna等。fasta文件中,第一行是由大于号">"开头的任意文字说明,用于序列标记,为了保证后续分析软件能够区分每条序列,单个序列的标识必须是唯一的,序列ID部分可以包含注释信息。从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号。序列部分可以在一行,也可以分成多行。
CSV文件是最通用的一种文件格式,它可以非常容易地被导入各种PC表格及数据库中。 此文件,一行即为数据表的一行。生成数据表字段用半角逗号隔开。
CSV是文本文件,用记事本就能打开,CSV (*.csv) 文件格式只能保存活动工作表中的单元格所显示的文本和数值。工作表中所有的数据行和字符都将保存。数据列以逗号分隔,每一行数据都以回车符结束。如果单元格中包含逗号,则该单元格中的内容以双引号引起。
原始fasta数据:
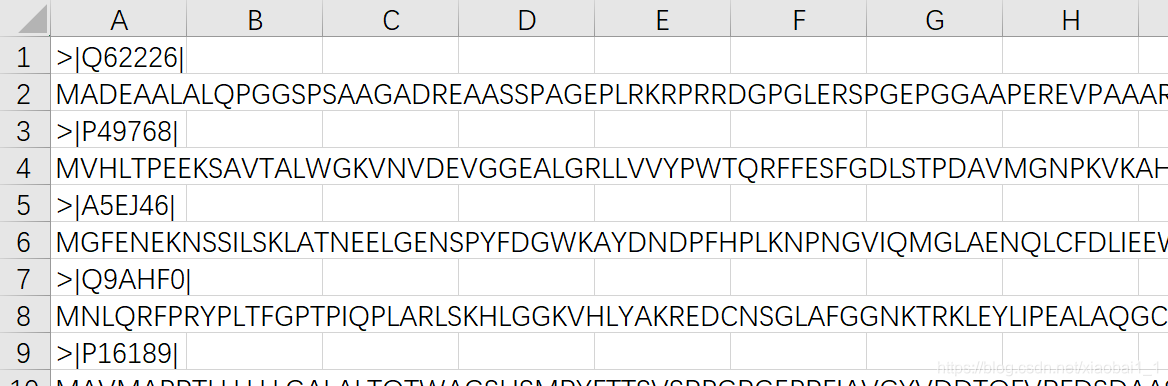
from Bio import SeqIO
import pandas as pd
meta = []
sequence = []
label=[]
i=0
seq = ('data.fasta') #转换的文件
for se 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 1057
1057











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









