







ChAMP加载肺癌数据和分析甲基化数据流程

1. 加载数据分析
#!/bin/evn R
rm(list = ls())
library(rstudioapi)
current_script_path <- getActiveDocumentContext()$path
# 将路径转换为当前目录
current_directory <- dirname(current_script_path)
# 设置当前工作目录
setwd(current_directory)
getwd()
library(ChAMP)
library(minfi)
library(Illumina450ProbeVariants.db)
library(sva)
library(IlluminaHumanMethylation450kmanifest)
library(limma)
library(RPMM)
library(DNAcopy)
library(preprocessCore)
library(impute)
library(marray)
library(wateRmelon)
library(goseq)
library(plyr)
library(GenomicRanges)
library(optparse)
library(pheatmap)
library(ggplot2)
filepath='D:/aR_project/methylation/extdata'
arraytype="450K"
out = "DNAmearray"
## 数据导入与过滤
data <- champ.load(filepath,arraytype = arraytype)
# 输出 data 的列名
names(data)
# [1] "beta" "intensity" "pd"
# 输出 data$beta 的维度
dim(data$beta)
# [1] 403116 8
# 检查是否存在缺失值
table(is.na(data$beta))
#
FALSE
3224928
# 输出 data$beta 的前几行
data$beta[1:2, 1:8]
C1 C2 C3 C4 T1 T2 T3 T4
cg00000957 0.7927427 0.8162839 0.8726977 0.8476140 0.8732848 0.8281842 0.6992366 0.7535339
cg00001349 0.6733450 0.6275007 0.6769735 0.7097567 0.4665632 0.7616798 0.2443499 0.4557010
# 输出 data$intensity 的维度
dim(data$intensity)
# [1] 403116 8
# 输出 data$pd 的维度
dim(data$pd)
# [1] 8 8
extdata文件夹内包含数据如下:
2. 数据质控
读取数据之后需要进行一些质控。
直接一个函数搞定:champ.QC()。
champ.QC(beta = data$beta,
pheno=data$pd$Sample_Group,
mdsPlot=TRUE,
densityPlot=TRUE,
dendrogram=TRUE,
PDFplot=TRUE,
Rplot=TRUE,
Feature.sel="None",
resultsDir="./")
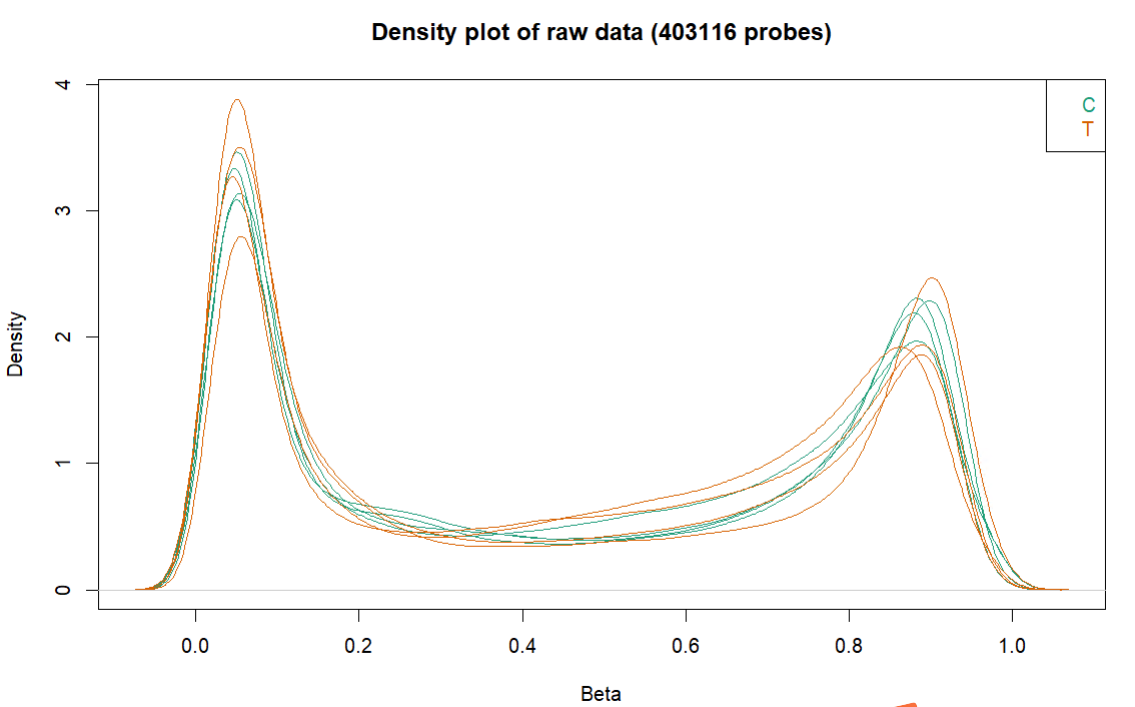
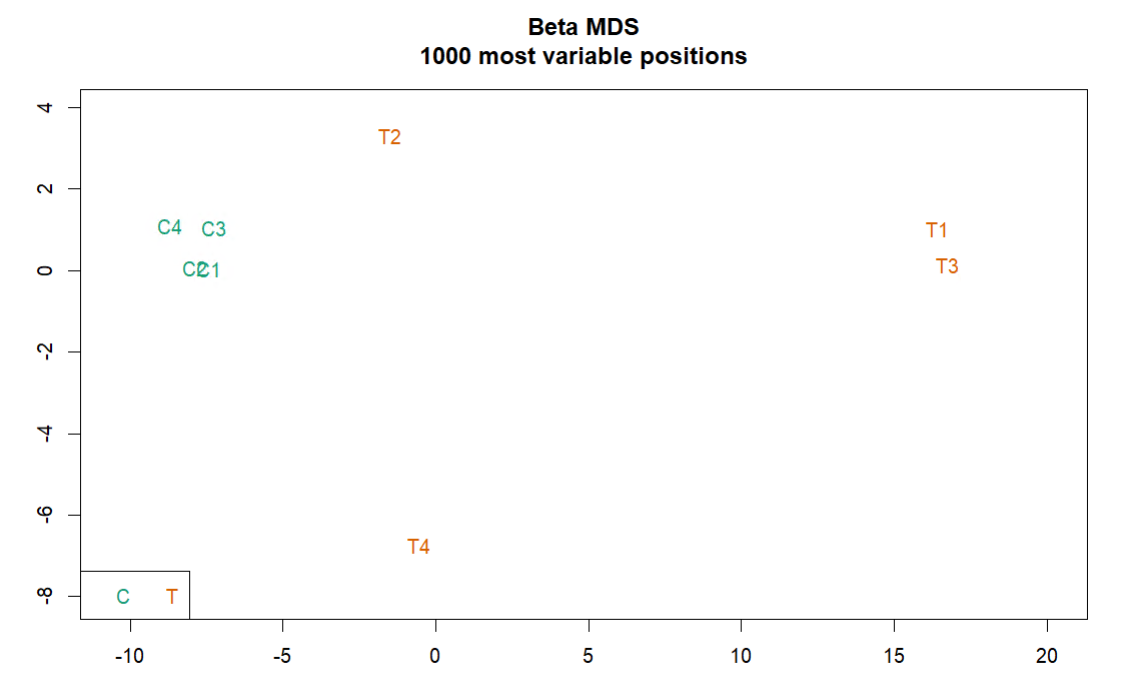
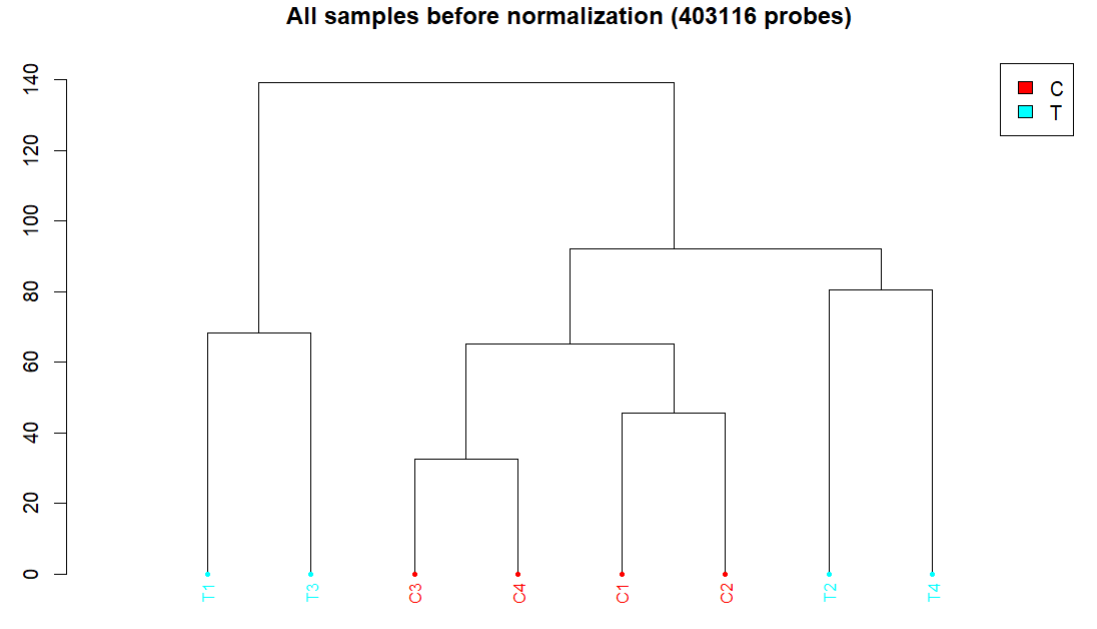
会生成3张图,放在CHAMP_QCimages这个文件夹下。
- MDS plot:根据前1000个变化最大的位点看样品相似性。
- densityPlot:每个样品的beta分布曲线,比较离群的可能是质量比较差的样本。
- 聚类图
3. 标准化
使用champ.norm()函数实现,提供4种方法:
- BMIQ,
- SWAN,
- PBC,
- FunctionalNormliazation
FunctionalNormliazation需要rgSet对象,SWAN需要rgSet和mset,PBC和BMIQ只需要beta 矩阵,FunctionalNormliazation和SWAN需要在读取数据时使用method = "minfi"。
myNorm <- champ.norm(beta = myLoad$beta,
arraytype = "EPIC",
cores = 8
)
3.1 方法解释
-
BMIQ (Beta Mixture Quantile Normalization)
-
用途:用于标准化 β 值,使其在各个样本之间具有相似的分布。
-
输入:只需要 β 值矩阵(
beta)。 -
使用方法
myNorm <- champ.norm(beta = myLoad$beta, method = "BMIQ", arraytype = "EPIC", cores = 8)
-
-
SWAN (Subset-quantile Within Array Normalization)
-
用途:考虑 Illumina BeadChip 上不同探针类型的差异,通过调整其分布来进行标准化。
-
输入:需要 β 值矩阵(
beta),以及 RGSet 和 MSet 对象。 -
使用方法
myNorm <- champ.norm(beta = myLoad$beta, rgSet = myLoad$rgSet, mset = myLoad$mset, me
-
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 7503
7503

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









