







关于甲基化的基础知识请自己学习了解,我们直接进行甲基化数据分析。
使用ChAMP包分析甲基化数据,这个包非常强大,甲基化数据分析必须得了解它。
这个包在bioconductor上,不要问我为什么install.packages("ChAMP")会失败,如果你有R包安装问题(包括任何R包安装问题),建议先看视频教程:
不过在使用ChAMP包的时候需要提供一个样本信息文件,.csv格式的,其实这个文件的准备非常简单。
ChAMP包给我们准备了一个示例数据集,我们先看一下示例数据是什么样的。
library("ChAMP")
testDir <- system.file("extdata",package="ChAMPdata")
在示例文件夹中的数据是这样的:
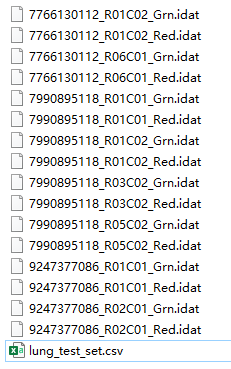
其中lung_test_set.csv就是样本信息,打开之后是这样的:
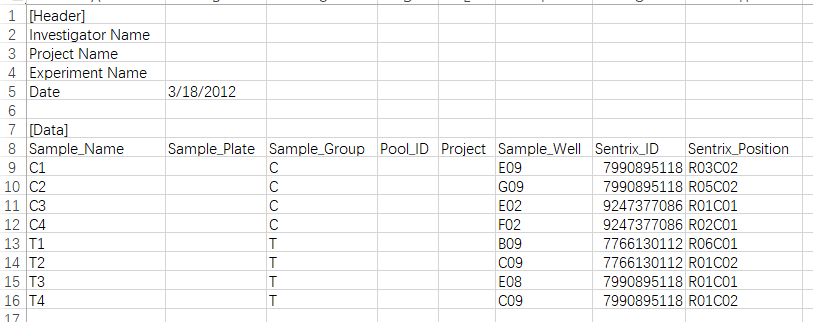
通常这个文件应该是作者给出的,但是很多时候都没有…
如果是你自己准备,需要注意以下几点:
- 这个csv文件中必须有的是
Sentrix_ID、Sentrix_Position,而且这两个名字不能改;这两列用_连接起来,就是你的IDAT文件的名字,而且必须用_连接,不然读取不了,所以你的IDAT文件的名字也必须得有_; Sample_Group这一列是分组信息(比如control和treat,normal和tumor等),没有这列信息也是可以读取的,但是没法做接下来的很多分析;这个名字可以改,但是在使用一些函数时也要跟着改;Sample_Name这一列是你的样本名字,没有这列信息也是可以读取的,但是有了更好,因为在过滤样本时要用;这个名字不能改;- 其余列可有可无,根据自己的需要来,比如你的数据如果有批次效应,你可以自己增加一列批次效应的信息,列名随便取,使用时指定即可;
- 上面示例csv文件中的前7行,没什么用,不需要;
- 这个文件夹中必须有且只有1个csv文件,文件名随便取。
如果没有Sample_Name这一列,就会出现下面的提示,所以Sample_Name这一列最好有,过滤样本时使用。Sample_Name这一列是你的样本名。
[ Section 1: Check Input Start ]
You have inputed beta,intensity for Analysis.
pd file provided, checking if it's in accord with Data Matrix...
!!! Your pd file does not have Sample_Name column, we can not check your Sample_Name, please make sure the pd file is correct.
如果你了解过minfi包,就会发现它们需要的这个文件是一样的,因为ChAMP读取这个数据是基于minfi包的。。。
下面用一个实际的例子来说明。
样本信息文件的制作
使用GSE149282这个数据集演示,一共24个样本,12个癌症,12个正常。下面这个分组信息可以直接从 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse149282 这个网页上复制粘贴即可,使用了之前介绍过的datapaste。
复制粘贴不走样的神奇R包,帮你快速从网页复制数据!
# 构建临床信息文件
# 粘贴过来稍加整理一下
pd <- data.frame(stringsAsFactors = FALSE,
Sample_Name = c("GSM4495491","GSM4495492","GSM4495493",
"GSM4495494","GSM4495495","GSM4495496",
"GSM4495497","GSM4495498","GSM4495499",
"GSM4495500","GSM4495501","GSM4495502",
"GSM4495503","GSM4495504","GSM4495505",
"GSM4495506","GSM4495507","GSM4495508",
"GSM4495509","GSM4495510","GSM4495511",
"GSM4495512","GSM4495513","GSM4495514"),
type = c("Colon adjacent normal 194T",
"Colon cancer tissue 194T",
"Colon adjacent normal 466T",
"Colon cancer tissue 466T",
"Colon adjacent normal 469T",
"Colon cancer tissue 469T",
"Colon adjacent normal 398T",
"Colon cancer tissue 398T",
"Colon adjacent normal 361T",
"Colon cancer tissue 361T",
"Colon adjacent normal 456T",
"Colon cancer tissue 456T",
"Colon adjacent normal 353T",
"Colon cancer tissue 353T",
"Colon adjacent normal 439T",
"Colon cancer tissue 439T",
"Colon cancer tissue 501T",
"Colon adjacent normal 501T",
"Colon cancer tissue 449T",
"Colon adjacent normal 449T",
"Colon cancer tissue 474T",
"Colon adjacent normal 474T",
"Colon cancer tissue 467T",
"Colon adjacent normal 467T")
)
我们需要把IDAT的文件名字加到pd中,并且把它们变成两列:Sentrix_ID和Sentrix_Position。
首先获取文件夹中IDAT文件名,后缀(_Red.idat/_Grn.idat)就不用了:
idat.name <- list.files("./gse149282/GSE149282_RAW/",
pattern = "*.idat.gz") |>
substr(1L,30L)
idat.name
## [1] "GSM4495491_200811050117_R01C01" "GSM4495491_200811050117_R01C01"
## [3] "GSM4495492_200811050117_R02C01" "GSM4495492_200811050117_R02C01"
## [5] "GSM4495493_200811050117_R03C01" "GSM4495493_200811050117_R03C01"
## [7] "GSM4495494_200811050117_R04C01" "GSM4495494_200811050117_R04C01"
## [9] "GSM4495495_200811050117_R05C01" "GSM4495495_200811050117_R05C01"
## [11] "GSM4495496_200811050117_R06C01" "GSM4495496_200811050117_R06C01"
## [13] "GSM4495497_200811050117_R07C01" "GSM4495497_200811050117_R07C01"
## [15] "GSM4495498_200811050117_R08C01" "GSM4495498_200811050117_R08C01"
## [17] "GSM4495499_200811050116_R01C01" "GSM4495499_200811050116_R01C01"
## [19] "GSM4495500_200811050116_R02C01" "GSM4495500_200811050116_R02C01"
## [21] "GSM4495501_200811050116_R03C01" "GSM4495501_200811050116_R03C01"
## [23] "GSM4495502_200811050116_R04C01" "GSM4495502_200811050116_R04C01"
## [25] "GSM4495503_200811050116_R05C01" "GSM4495503_200811050116_R05C01"
## [27] "GSM4495504_200811050116_R06C01" "GSM4495504_200811050116_R06C01"
## [29] "GSM4495505_200811050116_R07C01" "GSM4495505_200811050116_R07C01"
## [31] "GSM4495506_200811050116_R08C01" "GSM4495506_200811050116_R08C01"
## [33] "GSM4495507_202193490061_R01C01" "GSM4495507_202193490061_R01C01"
## [35] "GSM4495508_202193490061_R02C01" "GSM4495508_202193490061_R02C01"
## [37] "GSM4495509_202193490061_R03C01" "GSM4495509_202193490061_R03C01"
## [39] "GSM4495510_202193490061_R04C01" "GSM4495510_202193490061_R04C01"
## [41] "GSM4495511_202193490061_R05C01" "GSM4495511_202193490061_R05C01"
## [43] "GSM4495512_202193490061_R06C01" "GSM4495512_202193490061_R06C01"
## [45] "GSM4495513_202193490061_R07C01" "GSM4495513_202193490061_R07C01"
## [47] "GSM4495514_202193490061_R08C01" "GSM4495514_202193490061_R08C01"
把IDAT文件名添加到pd信息中去,由于我的这个例子中有两个_,所以你以任何一个_把它分为两部分都是可以的:
pd$Sentrix_ID <- substr(idat.name[seq(1,47,2)],1,23) #第一部分
pd$Sentrix_Position <- substr(idat.name[seq(1,47,2)],25,30) #第二部分
添加分组信息,我这个示例文件是分为normal和tumor两组:
pd$Sample_Type <- stringr::str_split(pd$type,pattern = " ",simplify = T)[,2]
pd[pd == "adjacent"] <- "normal"
pd <- pd[,-2]
这样这个pd样本信息文件就做好了:
head(pd)
## Sample_Name Sentrix_ID Sentrix_Position Sample_Type
## 1 GSM4495491 GSM4495491_200811050117 R01C01 normal
## 2 GSM4495492 GSM4495492_200811050117 R02C01 cancer
## 3 GSM4495493 GSM4495493_200811050117 R03C01 normal
## 4 GSM4495494 GSM4495494_200811050117 R04C01 cancer
## 5 GSM4495495 GSM4495495_200811050117 R05C01 normal
## 6 GSM4495496 GSM4495496_200811050117 R06C01 cancer
保存为CSV文件,文件名随便取即可:
write.csv(pd,file = "sample_type.csv",row.names = F,quote = F)
把这个csv文件和你的IDAT文件放在一个文件夹下就可以顺利读取了。
读取文件
# 加载R包
suppressMessages(library(ChAMP))
# 指定文件夹路径
myDir="./gse149282/GSE149282_RAW/"
## 我用的数据是Infinium MethylationEPIC,所以选EPIC,不要乱选哦!
myLoad <- champ.load(myDir, arraytype="EPIC")
OK,这样就非常顺利的读取成功了!
使用champ.load()函数可以直接读取IDAT文件(必须在同一个文件夹提供分组信息csv文件),在老版本的中这一步是借助minfi包实现的,但是现在默认是基于ChAMP方法实现的。主要不同是老版本的方法会同时返回rgSet和mset对象。这两种方法可以通过method参数切换。
champ.load()在读取数据时会自动过滤SNP,SNP信息主要来源于[这篇文章](Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of infinium dna methylation beadchip probes. Nucleic Acids Research. Published online 2016. doi:10.1093/nar/gkw967 “SNP参考信息”)。
champ.load()函数其实是champ.import()和champ.filter()两个函数的结合版,如果你想获得更加精细化的结果,需要使用两个函数。
如果你认真读上面的日志信息,也能发现champ.load()函数就是主要分为了两个步骤:champ.import()和champ.filter()。
首先是champ.import()部分:
第一件事:读取你的csv文件:
[ Section 1: Read PD Files Start ]
CSV Directory: ./gse149282/GSE149282_RAW/sample_type.csv
Find CSV Success
Reading CSV File
Replace Sentrix_Position into Array
Replace Sentrix_ID into Slide
There is NO Pool_ID in your pd file.
There is NO Sample_Plate in your pd file.
There is NO Sample_Well in your pd file.
[ Section 1: Read PD file Done ]
第二件事才是读取你的IDAT文件,在这一步还会给你很多提示信息:
[ Section 2: Read IDAT files Start ]
### 省略很多过程 ###
!!! Important !!!
Seems your IDAT files not from one Array, because they have different numbers of probe.
ChAMP wil continue analysis with only COMMON CpGs exist across all your IDAt files. However we still suggest you to check your source of data.
Extract Mean value for Green and Red Channel Success
Your Red Green Channel contains 1051815 probes.
[ Section 2: Read IDAT Files Done ]
第三步,添加注释信息:
[ Section 3: Use Annotation Start ]
Reading EPIC Annotation >>
Fetching NEGATIVE ControlProbe.
Totally, there are 411 control probes in Annotation.
Your data set contains 411 control probes.
Generating Meth and UnMeth Matrix
Extracting Meth Matrix...
Totally there are 865918 Meth probes in EPIC Annotation.
Your data set contains 865918 Meth probes.
Extracting UnMeth Matrix...
Totally there are 865918 UnMeth probes in EPIC Annotation.
Your data set contains 865918 UnMeth probes.
Generating beta Matrix
Generating M Matrix
Generating intensity Matrix
Calculating Detect P value
Counting Beads
[ Section 3: Use Annotation Done ]
然后是champ.filter()的部分。
在这一部分首先是check input,给出的信息可以说是太全面了!
[ Section 1: Check Input Start ]
You have inputed beta,intensity for Analysis.
pd file provided, checking if it's in accord with Data Matrix...
pd file check success.
Parameter filterDetP is TRUE, checking if detP in accord with Data Matrix...
detP check success.
Parameter filterBeads is TRUE, checking if beadcount in accord with Data Matrix...
beadcount check success.
parameter autoimpute is TRUE. Checking if the conditions are fulfilled...
!!! ProbeCutoff is 0, which means you have no needs to do imputation. autoimpute has been reset FALSE.
Checking Finished :filterDetP,filterBeads,filterMultiHit,filterSNPs,filterNoCG,filterXY would be done on beta,intensity.
You also provided :detP,beadcount .
[ Section 1: Check Input Done ]
check input之后就是过滤,根据你的input提供的信息进行各种过滤!
主要是6步过滤:
- First filter is for probes with detection p-value (default > 0.01).
- Second, ChAMP will filter out probes with ❤️ beads in at least 5% of samples per probe.
- Third, ChAMP will by default filter out all non-CpG probes contained in your dataset.
- Fourth, by default ChAMP will filter all SNP-related probes.
- Fifth, by default setting, ChAMP will filter all multi-hit probes.
- Sixth, ChAMP will filter out all probes located in chromosome X and Y.
Filtering probes with a detection p-value above 0.01.
Removing 7176 probes.
If a large number of probes have been removed, ChAMP suggests you to identify potentially bad samples
Filtering BeadCount Start
Filtering probes with a beadcount <3 in at least 5% of samples.
Removing 4832 probes
Filtering NoCG Start
Only Keep CpGs, removing 2953 probes from the analysis.
Filtering SNPs Start
Using general EPIC SNP list for filtering.
Filtering probes with SNPs as identified in Zhou's Nucleic Acids Research Paper 2016.
Removing 96223 probes from the analysis.
Filtering MultiHit Start
Filtering probes that align to multiple locations as identified in Nordlund et al
Removing 11 probes from the analysis.
Filtering XY Start
Filtering probes located on X,Y chromosome, removing 16458 probes from the analysis.
Updating PD file
Fixing Outliers Start
Replacing all value smaller/equal to 0 with smallest positive value.
Replacing all value greater/equal to 1 with largest value below 1..
[ Section 2: Filtering Done ]
All filterings are Done, now you have 738265 probes and 24 samples.
[<<<<< ChAMP.FILTER END >>>>>>]
上面所有的过滤都是可以通过参数控制的!真是太全面了!
需要注意,如果你只有beta matrix,champ.load()函数是不能执行某些过滤的,这时候你需要用champ.import()+champ.filter()。
myImport <- champ.import(myDir,)
myLoad <- champ.filter()
千万要注意这两个函数里面的参数选择哦!!
以上就是ChAMP包需要的样本信息csv文件的制作以及IDAT数据读取过程,下次继续!
之前学习了一下ChAMP包读取IDAT文件,真的是太贴心!
上次主要演示了ChAMP包需要的样本信息csv文件的制作以及IDAT数据读取过程。
今天继续走完后面的流程,很多日志文件我没放上来。
数据质控
读取数据之后需要进行一些质控。
直接一个函数搞定:champ.QC()。
champ.QC(beta = myLoad$beta,
pheno = myLoad$pd$Sample_Type # 注意列名要选对
)
会生成3张图,放在CHAMP_QCimages这个文件夹下。
- MDS plot:根据前1000个变化最大的位点看样品相似性。
- densityPlot:每个样品的beta分布曲线,比较离群的可能是质量比较差的样本。
- 聚类图

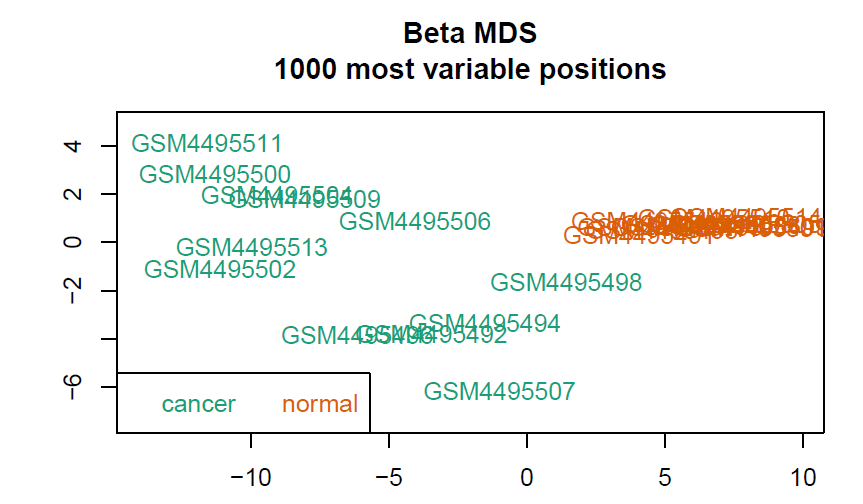
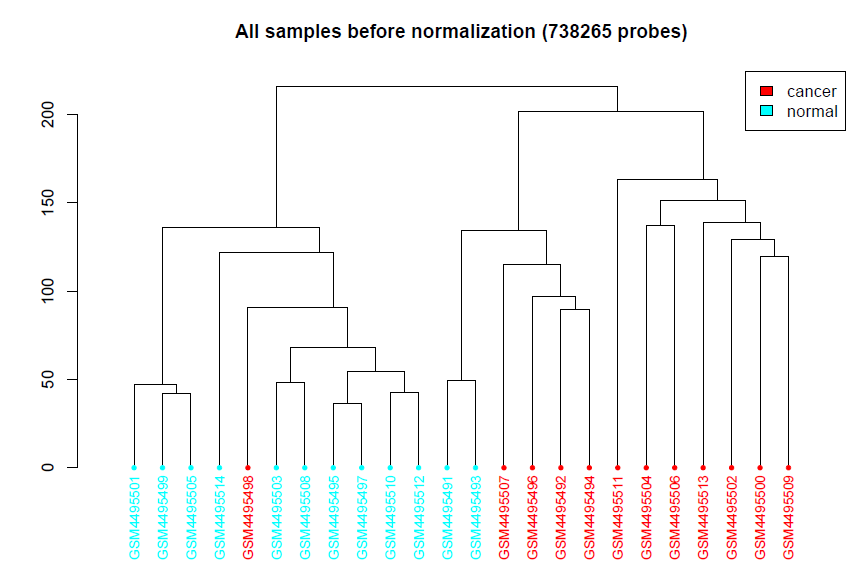
标准化
使用champ.norm()函数实现,提供4种方法:
- BMIQ,
- SWAN,
- PBC,
- FunctionalNormliazation
FunctionalNormliazation需要rgSet对象,SWAN需要rgSet和mset,PBC和BMIQ只需要beta 矩阵,FunctionalNormliazation和SWAN需要在读取数据时使用method = "minfi"。
myNorm <- champ.norm(beta = myLoad$beta,
arraytype = "EPIC",
cores = 8
)
SVD
奇异值分解。
可用于找出比较重要的变化。
champ.SVD(beta = myNorm |> as.data.frame(), # 这里需要注意
pd=myLoad$pd)
这个图中只有Sample_Type这个变量比较重要,如果你在csv文件中提供更多样本信息,这个图会看起来像热图。
save(myNorm,myLoad,file = "EPIC.rdata")
矫正批次效应
借助了sva包的Combat函数实现。
不是所有的都需要,根据自己的实际情况来。
注意使用时需要指定列名。
如果使用M值,需要指定logitTrans = T。
# 我们这个数据没有批次效应,就不运行这一步了
myCombat <- champ.runCombat(beta = myNorm,
pd = myLoad$pd,
batchname = c("Slide"), # pd文件中哪一列是批次信息
variablename = c("Sample_Type") # 指定分组列名,默认是Sample_Group
)
甲基化差异分析
甲基化有3个层次的差异分析:DMP,DMR,DMB:
- DMP代表找出Differential Methylation Probe(差异化CpG位点)
- DMR代表找出Differential Methylation Region(差异化CpG区域)
- Block代表Differential Methylation Block(更大范围的差异化region区域)
甲基化位点差异分析
DMP, Differentially Methylated Probes.
借助了limma包进行差异分析。支持多个分组的比较,如果分组大于2组,会自动进行两两比较。也支持数值型变量,比如年龄这种。
myDMP <- champ.DMP(beta = myNorm,
pheno = myLoad$pd$Sample_Type,
arraytype = "EPIC" # 别忘了改这里!!
)
结果就是myDMP[[1]],我们查看前6个,信息很多,一共20列:
head(myDMP[[1]])
logFC AveExpr t P.Value adj.P.Val B normal_AVG cancer_AVG deltaBeta CHR MAPINFO
cg16601494 -0.6402505 0.3910975 -28.66283 1.344757e-19 9.927870e-14 33.28137 0.07097231 0.7112228 0.6402505 1 1475737
cg09296001 -0.5763521 0.3706256 -27.71457 2.870274e-19 1.059511e-13 32.67560 0.08244952 0.6588016 0.5763521 7 127672564
cg22697045 0.3923163 0.6356132 25.87853 1.339179e-18 2.882866e-13 31.41759 0.83177142 0.4394551 -0.3923163 11 47359223
cg17301223 -0.6012424 0.4088559 -25.70149 1.561968e-18 2.882866e-13 31.28995 0.10823474 0.7094771 0.6012424 8 145106438
cg03241244 -0.5798955 0.3608224 -25.15421 2.529505e-18 3.734890e-13 30.88793 0.07087468 0.6507702 0.5798955 12 122017052
cg26256223 -0.5925507 0.3537249 -23.99254 7.275558e-18 7.140446e-13 29.99558 0.05744958 0.6500003 0.5925507 8 145106582
Strand Type gene feature cgi feat.cgi UCSC_Islands_Name SNP_ID SNP_DISTANCE
cg16601494 R I C1orf70 5'UTR shore 5'UTR-shore chr1:1476093-1476669 rs561063770 23
cg09296001 R I SND1 Body island Body-island chr7:127671158-127672853
cg22697045 F I MYBPC3 Body shore Body-shore chr11:47359953-47360216 rs3729950 21
cg17301223 R I OPLAH Body island Body-island chr8:145103285-145108027 rs566592895;rs148937107 51;47
cg03241244 F I KDM2B Body island Body-island chr12:122016170-122017693 rs112471261;rs531357599 21;33
cg26256223 R II OPLAH Body island Body-island chr8:145103285-145108027 rs553503071 20
还有图形化界面:
DMP.GUI(DMP = myDMP[[1]],
beta = myNorm,
pheno = myLoad$pd$Sample_Type
)
差异甲基化区域分析
Differentially Methylated Regions (DMRs)
myDMR <- champ.DMR(beta = myNorm,
pheno = myLoad$pd$Sample_Type,
arraytype="EPIC", # 注意选择!!
method = "Bumphunter"
)
提供图形化界面探索结果:
DMR.GUI(DMR=myDMR)
Differential Methylation Blocks
myBlock <- champ.Block(beta=myNorm,
pheno=myLoad$pd$Sample_Type,
arraytype="EPIC"
)
head(myBlock$Block)
chr start end value area cluster indexStart indexEnd L clusterL p.value fwer p.valueArea fwerArea
Block_1 5 142602343 171118422 0.1663742 512.5989 74 225817 228897 3081 8726 0 0 0.000000e+00 0.000
Block_2 12 73523354 112200054 0.1249276 496.5871 168 67195 71169 3975 7196 0 0 0.000000e+00 0.000
Block_3 2 18435 25360029 0.1308144 423.4463 20 137105 140341 3237 10107 0 0 0.000000e+00 0.000
Block_4 7 22371030 43698429 0.1640385 418.1342 92 250282 252830 2549 4868 0 0 0.000000e+00 0.000
Block_5 11 118313315 134945796 0.1556909 405.2634 161 56593 59195 2603 4782 0 0 2.413494e-06 0.002
Block_6 2 48641899 85085187 0.1250336 401.1079 20 143636 146843 3208 10107 0 0 2.413494e-06 0.002
同样是提供图形化界面查看结果:
Block.GUI(Block=myBlock,
beta=myNorm,
pheno=myLoad$pd$Sample_Type,
runDMP=TRUE,
compare.group=NULL,
arraytype="EPIC")
save(myDMP,myDMR,myBlock,file = "./gse149282/gse149282_dmprb.rdata")
富集分析
提供GSEA分析的函数,这一步完全可以使用更加专业的clusterprofiler。
myGSEA <- champ.GSEA(beta = myNorm,
DMP = myDMP[[1]],
DMR = myDMR,
arraytype = "EPIC",
adjPval = 0.05,
method = "gometh"
)
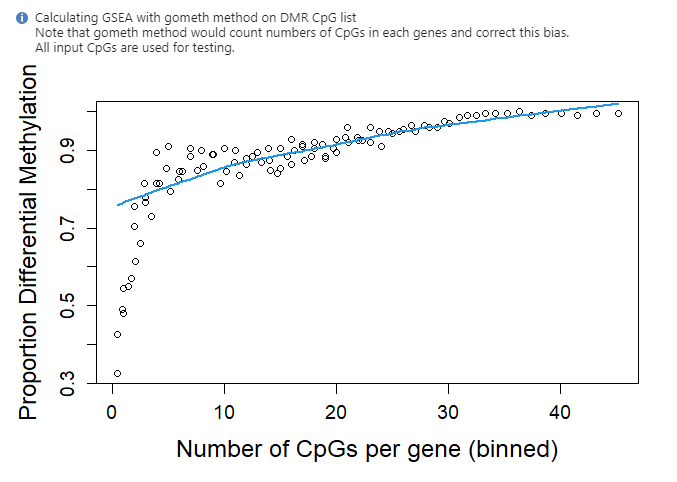
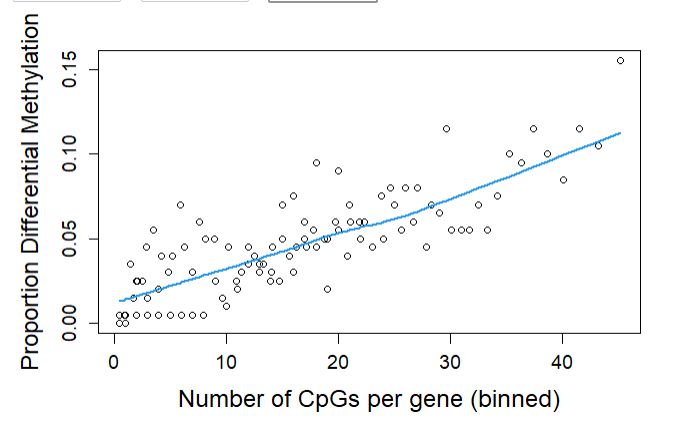
head(myGSEA$DMP)
ONTOLOGY TERM N DE P.DE FDR
GO:0071944 CC cell periphery 5604 5332 1.072641e-74 2.430284e-70
GO:0005886 CC plasma membrane 5139 4885 1.605572e-65 1.818872e-61
GO:0032501 BP multicellular organismal process 6970 6478 2.138994e-40 1.615440e-36
GO:0007154 BP cell communication 6026 5607 3.068516e-35 1.691964e-31
GO:0031224 CC intrinsic component of membrane 5082 4739 3.733865e-35 1.691964e-31
GO:0016020 CC membrane 8634 7958 6.456365e-35 2.438031e-31
head(myGSEA$DMR)
ONTOLOGY TERM N DE P.DE FDR
GO:0003700 MF DNA-binding transcription factor activity 1336 195 1.108904e-25 2.512443e-21
GO:0000981 MF DNA-binding transcription factor activity, RNA polymerase II-specific 1290 189 6.300238e-25 7.137224e-21
GO:0000977 MF RNA polymerase II transcription regulatory region sequence-specific DNA binding 1334 190 3.119916e-23 2.356265e-19
GO:1990837 MF sequence-specific double-stranded DNA binding 1479 200 2.737843e-21 1.550783e-17
GO:0000976 MF transcription cis-regulatory region binding 1426 193 1.390539e-20 6.301089e-17
GO:0001067 MF transcription regulatory region nucleic acid binding 1428 193 1.801922e-20 6.804360e-17
还可以用贝叶斯方法,这种方法不需要DMP或者DMR,只要提供β矩阵和分组信息即可:
myebayGSEA <- champ.ebGSEA(beta = myNorm,
pheno = myLoad$pd$Sample_Type,
arraytype = "EPIC"
)
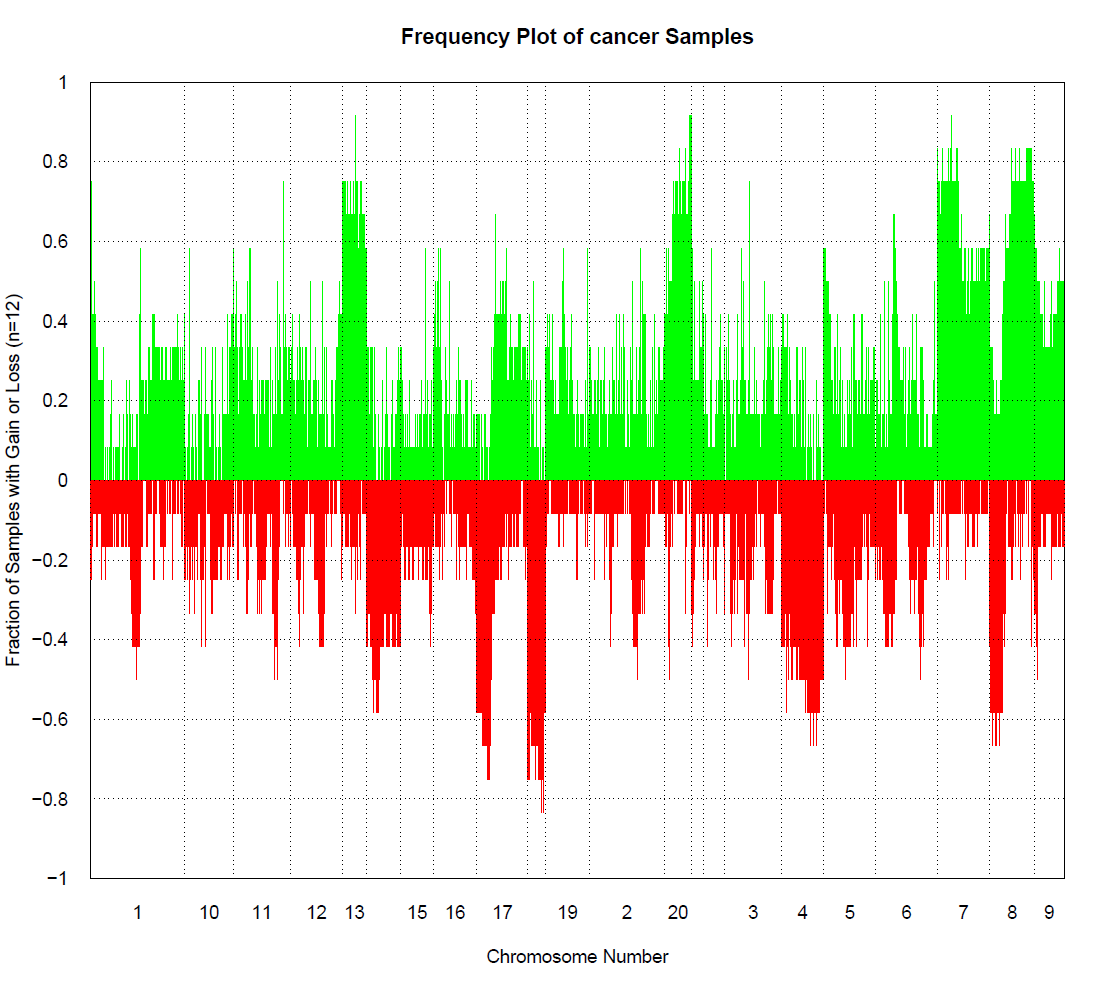
拷贝数变异分析
借助HumanMethylation450/HumanMethylationEPIC data实现这个功能,所以可能不能分析27K数据。
提供2种方法分析拷贝数变异。一种是分析两个状态间的拷贝数差异,比如说normal和cancer,另一种是分析单个样本的拷贝数和总体平均拷贝数。
使用第一种方法需要指定和谁比,如果是血液样本,这个包自带了一些血液样本可以比较,不需要指定controlGroup,但是如果不是血液样本,需要指定和谁比。
使用第二种方法只要选择control=FALSE即可。
# 非常耗时!
myCNA <- champ.CNA(intensity = myLoad$intensity,
pheno = myLoad$pd$Sample_Type,
arraytype = "EPIC",
controlGroup = "normal" # 指定和normal比
)
这个过程很慢,会在CHAMP_CNA文件夹中生成很多图。
标准化流程就是这么多,在ChAMP中都是一个函数搞定,基因注释等都是自动完成的,太方便了!
EPIC数据的甲基化分析在ChAMP中非常简单,就是这几步:
# 数据读取
myDir="./gse149282/GSE149282_RAW/"
myLoad <- champ.load(myDir, arraytype="EPIC")
# 数据预处理
champ.QC()
myNorm <- champ.norm(arraytype="EPIC")
champ.SVD()
myCombat <- champ.runCombat() # 可选
# 差异分析
myDMP <- champ.DMP(arraytype="EPIC")
myDMR <- champ.DMR(arraytype="EPIC")
myBlock <- champ.Block(arraytype="EPIC")
# 富集分析
myGSEA <- champ.GSEA(arraytype="EPIC")
# 拷贝数分析
myCNA <- champ.CNA(arraytype = "EPIC")
450K的数据也是一模一样的流程!
上面所有的步骤,这个包还提供了一个一步法完成:
champ.process(directory = myDir)
不过还是分步运行更能获得自己想要的结果哦。

















 1046
1046

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









