






gff是以 "\t" 为分隔符的存储9列信息的文本文件,格式如下:
chr1H PGSBv2.28112019 gene 222337 227461 3053.527 + . ID=Horvu_AKASHIN_1H01G000500;OGID=OG0028375;AHRD Descrip
tion=Fatty acyl-CoA reductase;AHRD Quality Score=3;TE-related=0;PFAM ID=PF03015,PF07993;GO ID=GO:0006629,GO:0016020,GO:0016021,GO:001649
1,GO:0055114,GO:0080019,GO:0102965
chr1H PGSBv2.28112019 mRNA 222337 227461 . + . ID=Horvu_AKASHIN_1H01G000500.1;Parent=Horvu_AKASHIN_1H01G000500
chr1H PGSBv2.28112019 exon 222337 222427 . + . ID=Horvu_AKASHIN_1H01G000500.1_exon_1;Parent=Horvu_AKASHIN_1H01G000500.1
chr1H PGSBv2.28112019 CDS 222337 222427 . + 0 Parent=Horvu_AKASHIN_1H01G000500.1seqid:参考序列的id。source:注释的来源。如果未知,则用点(.)代替。一般指明产生此gff3文件的软件或方法。type: 类型,此处的名词是相对自由的,建议使用符合SO惯例的名称(sequenceontology),如gene,repeat_region,exon,CDS等。start:开始位点,从1开始计数(区别于bed文件从0开始计数)。end:结束位点。score:得分,对于一些可以量化的属性,可以在此设置一个数值以表示程度的不同。如果为空,用点(.)代替。strand:“+”表示正链,“-”表示负链,“.”表示不需要指定正负链。phase:步进。对于编码蛋白质的CDS来说,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过的碱基个数。attributes:属性。一个包含众多属性的列表,格式为“标签=值”(tag=value),不同属性之间以分号相隔。
要求:从gff中提取cDNA长度和exon数量
思考:cDNA是由mRNA反转录得到的,故而求mRNA长度即可;在文件中对于一个基因的信息而言,总是按gene、mRNA、exon、CDS的顺序出现;
将文件f导入后按行读取,以 "\t" 分割成列表,
mRNA的长度只要判断第三列( f[2] )为mRNA后,提取位置信息,长度=结尾-开始+1
exon个数可以计数获得,构建字典,键值设为ID,当(读到CDS或者)读到下一个基因时停止并重新计数
开始想同时进行输入和输出,计算量很大,易出错:
# 提取mRNA信息和exon数量,同时进行
import os
dirs="/data/database/barley_pan_genome/barley_genome/gtf"
files=os.listdir(dirs) #列出该目录下的所有文件
name=open("/data/user/ZY/ZY/NLR/1AKashinriki/location/name","r")
zd1={}
zd2={}
list=[]
#首先20个品种共7450个基因依次在20个gff文件中进行循环
for l in name.readlines():
l=l.replace("\n","") #需要把换行符替换掉,exon行不包含换行符
for f in files:
gtf=open("/data/database/barley_pan_genome/barley_genome/gtf/{q}".format(q=f),"r")
i=0 #exon计数可用,也可放置在上个循环内
for lines in gtf.readlines():
a=lines.split("\t")
#1、如果id与品种的gff文件不匹配,在遍历完gff所有行后会进行下一个文件,直至匹配到相同品种的gff
#2、如果id与品种的gff文件匹配:
#1)目标id与行中的id匹配,且第三列为mRNA,那么计算长度
#2)id匹配且为exon,i+1,
# 当id匹配,i不为0,且类型为CDS时,输出exon个数后,跳出第二个循环,直接换个id重新循环
#3)当id不匹配时,继续循环
if l in a[8]and a[2] == "mRNA":
le=int(a[4])-int(a[3])+1
#out内储存mRNA长度相关信息
out=open("/data/user/ZY/ZY/NLR/length/all.csv","a+")
out.write(a[3]+","+a[4]+","+str(le)+","+a[8])
out.close()
elif l in a[8] and a[2] == "exon":
i += 1
elif l in a[8] and a[2] == "CDS" and i != 0:
with open("/data/user/ZY/ZY/NLR/length/all.csv", "r") as input:
file=input.readlines()
newline = file[-1] #输出时选取all文件的最后一列,但前提是该文件不能为空
newout = open("/data/user/ZY/ZY/NLR/length/all_exon.csv","a+")
newout.write(newline+","+str(i))
input.close()
break #跳出当前循环
else:
continue
break参考凌一民的脚本后进行修改,提取信息和输出信息分开进行,计算量小很多,但是需要进行20次提交命令
先提取需要的信息存到字典当中,字典的键都是id,然后写个小循环输出
'''
获取gff中相关基因的信息,提取mRNA长度,exon数量,
chr1H PGSBv2.28112019 gene 222337 227461 3053.527 + . ID=Horvu_AKASHIN_1H01G000500;OGID=OG0028375;AHRD Descrip
tion=Fatty acyl-CoA reductase;AHRD Quality Score=3;TE-related=0;PFAM ID=PF03015,PF07993;GO ID=GO:0006629,GO:0016020,GO:0016021,GO:001649
1,GO:0055114,GO:0080019,GO:0102965
chr1H PGSBv2.28112019 mRNA 222337 227461 . + . ID=Horvu_AKASHIN_1H01G000500.1;Parent=Horvu_AKASHIN_1H01G000500
chr1H PGSBv2.28112019 exon 222337 222427 . + . ID=Horvu_AKASHIN_1H01G000500.1_exon_1;Parent=Horvu_AKASHIN_1H01G000500.1
chr1H PGSBv2.28112019 CDS 222337 222427 . + 0 Parent=Horvu_AKASHIN_1H01G000500.1
'''
import sys
length={}
NLR_length={}
exon_number={}
NLR_exon_number={}
gff=open(sys.argv[1],"r")
name=open("/data/user/ZY/ZY/NLR/1AKashinriki/location/name","r")
# for n in name:
# exon=0 不能同时进行基因名称的循环
exon = 0
#te = "Horvu_AKASHIN_1H01G000100" 方法二,对exon清零
for line in gff:
if line.startswith("#"):
continue
col=line.split("\t")
id=col[8].split(";")[0].replace("ID=","")
id=id[:25]
type=col[2]
start=int(col[3])
end=int(col[4])
strand=col[6]
if type == "mRNA":
len_mRNA=end-start+1
length[id]=len_mRNA
if type == "exon":
#exon += 1
if id not in exon_number.keys():
exon_number.update({id: 0})
exon_number[id] += 1
# if te != id:
# te = id
# exon = 0
gff.close()
for n in name:
n=n.replace("\n","")
if n in length.keys():
NLR_length[n]=length[n]
NLR_exon_number[n]=exon_number[n]
out=open("finaly.csv","a+")
out.write(str(NLR_length[n])+","+str(NLR_exon_number[n])+","+n+"\n")
out.close()
print("文件循环结束")
接下来加入pep_length,从已经得到的20个品种NLR基因的pep.fasta文件中获得
思路:先匹配到 ">" 那一行,处理后得到id,作为key;将下一行长度作为值
外部要写20个文件名,有点麻烦
#计算蛋白序列长度
import sys
import pandas as pd
sequence={}
for i in range(20):
i += 1
pep = open(sys.argv[i])
#sequence = {}
for line in pep.readlines():
line = line.strip() #去掉line左右两端空格
if line.startswith(">"):
name = line[1:]
sequence[name] = ''
else:
sequence[name] = len(line)
pep.close()
#需要在finaly文件中加列名
pep_length=sequence.values()
df = pd.read_csv("/data/user/ZY/ZY/NLR/length/finaly.csv")
# df.columns=["mRNA_length","exon_num","ID"],不能这么加列名
df["pep_length"]=pep_length
df.to_csv("/data/user/ZY/ZY/NLR/length/finaly2.csv",index=False,sep=",")再查PI值
查PI值网站:ExPASy - Compute pI/Mw tool![]() https://web.expasy.org/compute_pi/
https://web.expasy.org/compute_pi/
需要提交的是序列信息或者Swiss-Prot的id,本想在g:Covert转化,转化不成功,只能用序列

先将所有蛋白序列删除 ">" 所在行后提取至一个文件中,然后消除文件中的 *号,提交到网页上,等待结果。结果出来后选择可导出模式,复制到文本文件中,用python进行处理。

获得的文本文件格式内容如下:
PI所在行含有 . ,且可按 "\t" 分割
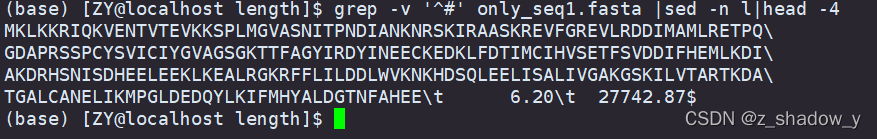
查看文件分隔符命令:(内为小写L)
grep -v "^#" 文件名 | sed -n l | head
#提取PI
import pandas as pd
info=open(r"D:\张颖\NLR\base_info\only_seq1.fasta","r")
PI=[]
for line in info.readlines():
if "." in line:
pi = line.split("\t")[1].strip() #strip()删除空格
PI.append(pi)
info.close()
#用pandas打开文件时,路径不能有中文
v=open(r"D:\张颖\NLR\base_info\finaly2.csv","r")
df=pd.read_csv(v)
df["PI"]=PI
f=open(r"D:\张颖\NLR\base_info\finaly3.csv","w")
df.to_csv(f,index=False,sep=",")至此,基因的cDNA长度,exon数量,蛋白质长度,等电点信息全部提取完成。















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









