







 本文详细介绍了开源工具RDKit在化学信息学中的应用,涉及安装、读写分子操作(如SMILES/SDF)、分子可视化以及3D展示,展示了如何使用RDKit进行化合物描述符生成和分子结构分析。
本文详细介绍了开源工具RDKit在化学信息学中的应用,涉及安装、读写分子操作(如SMILES/SDF)、分子可视化以及3D展示,展示了如何使用RDKit进行化合物描述符生成和分子结构分析。
🌞欢迎来到AI+医学的世界
🌈博客主页:卿云阁💌欢迎关注🎉点赞👍收藏⭐️留言📝
🌟本文由卿云阁原创!
📆首发时间:🌹2024年3月17日🌹
✉️希望可以和大家一起完成进阶之路!
🙏作者水平很有限,如果发现错误,请留言轰炸哦!万分感谢!
目录
RDKit 介绍
化合物(compounds)药物是一种主要的药物类型,通常进行相关研究时通常需要对其结构进行操作,展示,及分子量,化学描述符等计算。这里介绍一个操作简便友好的开源工具,RDKit。
RDKit是一个用于化学信息学的开源工具包,基于对化合物2D和3D分子操作,利用机器学习方法进行化合物描述符生成,fingerprint生成,化合物结构相似性计算,2D和3D分子展示等。基于PYTHON语言进行调取使用。
该工具官网:https://rdkit.org
也可以根据从 The RDKit Documentation 网站进入了解RDKit并熟悉RDKit的指令操作。
以下介绍RDKit的一些基础操作。
RDKit安装
这里介绍一种最快速的安装方法,由于RDKit是基于python语言使用的,所以可以在anaconda上快速进行RDKit的安装。
pip install -i https://pypi.tuna.tsinghua.edu.cn/simple rdkit-pypi安装完成后,可以在python界面进行import,来check RDKit 是否安装成功。如果可能顺利import,则说明安装成功。
import rdkit
读分子操作
RDKit 支持从Smiles、mol、sdf 文件中读入分子获得分子对象。 |Smiles、mol 是通常用于保存单个分子;而sdf格式当初是作为分子库形式设计的。 因此读入sdf得到的是分子迭代器,读入Smiles 和mol 文件是分子对象。
SMILES简介
SMARTS(SMiles ARbitrary Target Specification)是一种用于描述分子模式和属性的语言。SMILES所有的符号和属性在SMARTS中同样适用,因此它也是SMILES的延伸。此外,SMARTS还包括了逻辑操作符和额外的分子描述符,后文会一一介绍。
(1)从SMILES/SMARTS直接读取
smi='CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3': 这行代码定义了一个字符串变量smi,其中包含了一个SMILES表示的化合物的信息。SMILES(Simplified Molecular Input Line Entry System)是一种用于表示分子结构的文本字符串表示法。
mol = Chem.MolFromSmiles(smi): 这行代码使用RDKit的MolFromSmiles()函数将SMILES字符串转换为RDKit的分子对象。MolFromSmiles()函数将SMILES字符串解析并创建一个分子对象mol。
sms = Chem.MolFromSmarts('Cc1ccccc1'): 这行代码使用RDKit的MolFromSmarts()函数将SMARTS模式字符串转换为RDKit的分子对象。SMARTS(SMILES Arbitrary Target Specification)是一种类似于SMILES的表示法,用于描述分子的子结构模式。
print(mol): 这行代码打印了分子对象mol的信息,通常包括分子的原子、键和立体化学信息。
print(sms): 这行代码打印了分子对象sms的信息,通常包括SMARTS模式的表示信息。<rdkit.Chem.rdchem.Mol object at 0x0000025E71FBC970> <rdkit.Chem.rdchem.Mol object at 0x0000025E730D1120>
<rdkit.Chem.rdchem.Mol object at 0x0000025E71FBC970>和<rdkit.Chem.rdchem.Mol object at 0x0000025E730D1120>表示了两个分子对象的内存地址,而不是它们的化学结构。要打印出分子的化学结构,你可以使用RDKit提供的其他方法来获得分子的具体信息,比如通过Draw模块来绘制化学结构。(2)文件批量读取
- 从.smi批量读取:SmilesMolSupplier(data, delimiter, smilesColumn, nameColumn, titleLine, sanitize)
data:数据文件
delimiter:分隔符,默认为' '
smilesColumn:SMILES所在列,默认为0
nameColumn:SMILES名称所在列,默认为1
titleLine:是否含有标题行,默认True
sanitize:是否检查正确性,默认Truesuppl = Chem.SmilesMolSupplier('data/batch_smiles.smi', delimiter='\t') mols = [Chem.MolToSmiles(mol) for mol in suppl] print(mols)['C1=CC=CC=CC=C1', 'c1ccccc1', 'c1ccoc1']
(3)文本批量读取
- 从文本批量读取SmilesMolSupplierFromText()
参数基本同上with open('data/batch_smiles.smi', 'r') as f: mols_text = f.read() suppl = Chem.SmilesMolSupplierFromText(mols_text, delimiter='\t') mols = [Chem.MolToSmiles(mol) for mol in suppl] print(mols)['C1=CC=CC=CC=C1', 'c1ccccc1', 'c1ccoc1'](4)DataFrame批量读取
- 读取DataFrame中的SMILES:PandasTools.AddMoleculeColumnToFrame(frame, smilesCol, molCol, includeFingerprints)
frame:DataFrame对象
smilesCol:SMILES所在列
molCol:新列名,将存放产生的rdkit mol对象
includeFingerprints:是否生成指纹df = pd.read_csv('data/smiles_df.csv') PandasTools.AddMoleculeColumnToFrame(df,'SMILES','mol',includeFingerprints=True) df['MW'] = df['mol'].apply(Descriptors.MolWt) print(df.head(2))
读.sdf
文件批量读取
- 从.sdf里批量读取:Chem.SDMolSupplier(fileName, sanitize, removeHs, strictParsing)
fileName:文件名
sanitize:检查化合价,计算芳香性、共轭、杂化、kekule,默认True
removeHs:是否隐藏氢原子,默认True
strictParsing:是否使用严格模式进行解析,默认Truesuppl = Chem.SDMolSupplier('data/batch.sdf') mols = [Chem.MolToSmiles(mol) for mol in suppl if mol] print(mols)['C1=C\\C=C/C=C\\C=C/1', 'c1ccccc1', 'c1ccoc1']压缩包批量读取
gz_file = gzip.open('data/batch.sdf.gz', 'r') suppl = Chem.ForwardSDMolSupplier(gz_file) mols = [Chem.MolToSmiles(mol) for mol in suppl if mol] print(mols) f.close()['C1=C\\C=C/C=C\\C=C/1', 'c1ccccc1', 'c1ccoc1']读.mol
m = Chem.MolFromMolFile('data/output.mol') print(Chem.MolToSmiles(mol))c1cocc1
写分子操作
RDKit 可以把分子对象保存成Smiles、molBlock、mol、inchi、inchikey文件。
输出二进制
- 可以使用python的pickle将分子转成二进制
相对于SMILES和文件,二进制pkl格式体积更小,读取速度更快- 输出pkl文件:pickle.dump(obj, file, ...)
obj:要封装的对象
file:要写入的文件m = Chem.MolFromSmiles('c1ccncc1') with open('data/output.pkl', 'wb') as f: pickle.dump(m, f)pkl = pickle.dumps(m) pkl
binStr = m.ToBinary() binStr
输出SMILES/SMARTS
输出默认式
- 输出SMILES:MolToSmiles(mol, isomericSmiles, kekuleSmiles, canonical, ...)
kekuleSmiles:默认False,不使用kekule时:脂肪族碳用"C"表示(大写),芳香族用"c"表示(小写)
isomericSmiles:默认True,区分同分异构体("@"表示手性,""和"/"表示顺反异构)
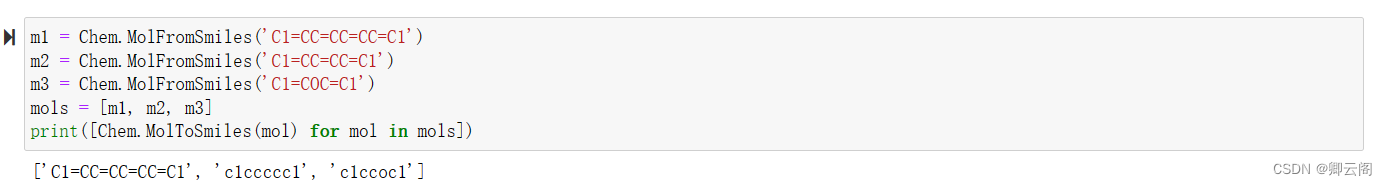
canonical:默认True,输出标准SMILESm1 = Chem.MolFromSmiles('C1=CC=CC=CC=C1') m2 = Chem.MolFromSmiles('C1=CC=CC=C1') m3 = Chem.MolFromSmiles('C1=COC=C1') mols = [m1, m2, m3] print([Chem.MolToSmiles(mol) for mol in mols])
输出.sdf
批量输出到.sdf
- 批量输出到文件:SDWriter()
使用方法类似于SMILES的批量输出
可以自定义属性信息,并记录在.sdf文件中
返回writer对象- 写入sdf:writer(mol, confId)
mol:mol对象
conFId:写入的第几个构象(不同构象坐标不一样)writer = Chem.SDWriter('data/batch.sdf') writer.SetProps(['LOGP', 'MW']) for i, mol in enumerate(mols): mw = Descriptors.ExactMolWt(mol) logp = Descriptors.MolLogP(mol) mol.SetProp('MW', '%.2f' %(mw)) mol.SetProp('LOGP', '%.2f' %(logp)) mol.SetProp('_Name', 'No_%s' %(i)) writer.write(mol) writer.close()
分子可视化
单个展示
- 从mol对象到图片:MolToImage(mol, size, kekulize, wedgeBonds, fitImage, ...)
mol:mol对象
size:图片尺寸,默认(300, 300)
kekulize:是否展示kekule形式,默认True(True:芳香系统用实线表示,False:虚线表示)
wedgeBonds:是否展示楔形键,即立体构型,默认Truemol = Chem.MolFromSmiles('C[C@H](O)c1ccccc1') Draw.MolToImage(mol, size=(150,150), kekulize=True)
保存图片
MolToFile(mol, filename, size, kekulize, wedgeBonds, ...)参数基本同上
Draw.MolToFile(mol, 'data/output.png', size=(150, 150))批量展示
从DataFrame中展示
- 从df中展示:FrameToGridImage(frame, column, molsPerRow, subImgSize, legendsCol, ...)
frame:DataFrame对象
column:rdkit mol对象所在列
molsPerRow,:每行显示的分子数
subImgSize:图片大小
legendsCol:标题所在列df = pd.read_csv('data/smiles_df.csv') PandasTools.AddMoleculeColumnToFrame(df,'SMILES','mol',includeFingerprints=True) img = PandasTools.FrameToGridImage(df, column='mol', molsPerRow=5, subImgSize=(200,200), legendsCol='Name') img
保存
img.save('data/df.png')3D展示
转换3D时,为了得到靠谱的三维构象,一般先加氢:AddHs(mol)
通过距离几何算法计算3D坐标:EmbedMolecule(mol, randomSeed, ...)
mol:mol对象
randomSeed:随机种子转换完后再进行一步力场优化,比如MMFF94:MMFFOptimizeMolecule(mol)
m3d = Chem.MolFromSmiles('CNC(=O)N(N(CCCl)S(C)(=O)=O)S(C)(=O)=O') m3d = Chem.AddHs(m3d) AllChem.EmbedMolecule(m3d, randomSeed=3) AllChem.MMFFOptimizeMolecule(m3d)Draw.MolToImage(m3d, size=(250,250))
常用功能
读取分子
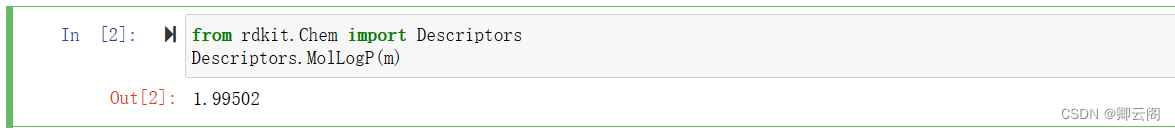
from rdkit import Chem from rdkit.Chem import AllChem m = Chem.MolFromSmiles('Cc1ccccc1') #创建一个分子,生成RDMol对象 smi = Chem.MolToSmiles(m)RDKit描述符
from rdkit.Chem import Descriptors Descriptors.MolLogP(m)
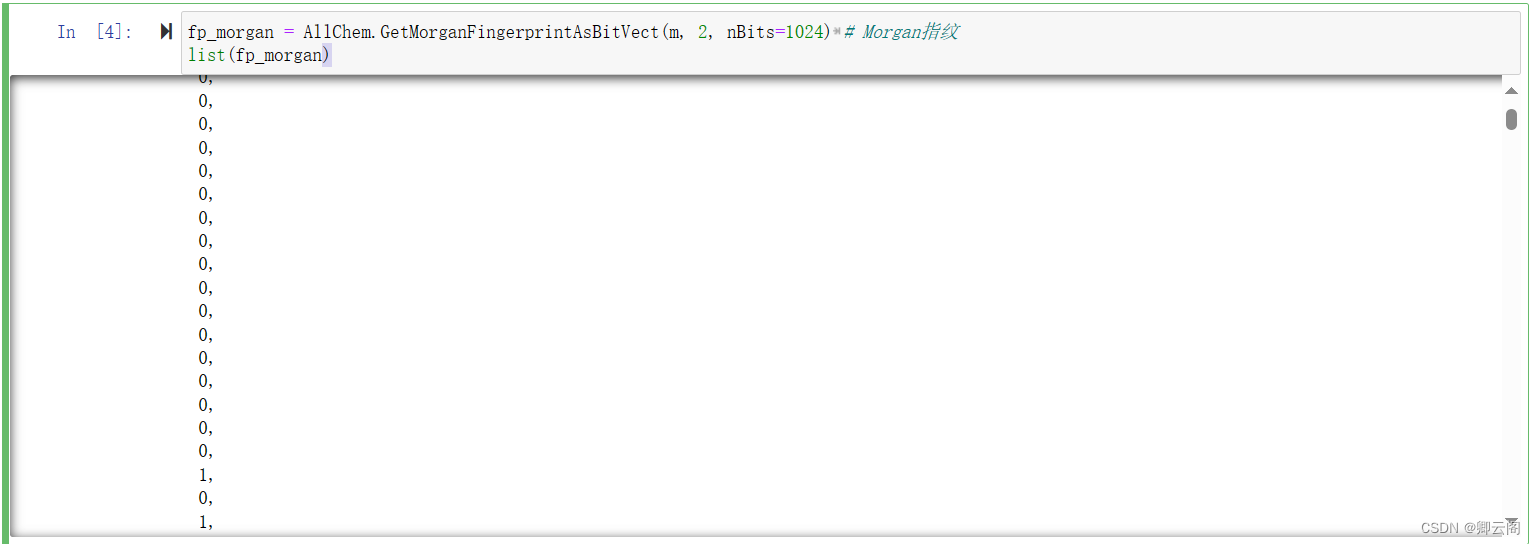
生成分子指纹
fp_morgan = AllChem.GetMorganFingerprintAsBitVect(m, 2, nBits=1024) # Morgan指纹 list(fp_morgan)





















01-15
 405
405
405
07-06
2427
2427
06-29
7407
7407

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











