







一、从GEO官网下载表达和临床数据
GEO数据库构成:
1. GEO Platform(GPL)
-
定义:描述实验使用的芯片或测序技术平台。
-
内容:包含探针与基因的对应关系、平台设计参数等元数据。
-
示例:
GPL570(Affymetrix Human Genome U133 Plus 2.0 Array)。
2. GEO Sample(GSM)
-
定义:单个生物样本的实验数据。
-
内容:原始数据(如CEL文件)、处理后的表达矩阵及样本描述(如疾病状态、处理条件)。
-
示例:
GSM12345(肺癌组织样本的基因表达谱)。
3. GEO Series(GSE)
-
定义:关联多个样本(GSM)的完整研究项目。
-
内容:实验设计、分析方法及所有相关GSM和GPL的索引。
-
示例:
GSE12345(包含20个样本的肺癌转录组研究)。
4. GEO Dataset(GDS)
-
定义:经NCBI整合和标准化的数据集。
-
内容:统一格式的表达矩阵、实验注释及差异分析工具。
-
示例:
GDS1234(标准化后的乳腺癌基因表达数据集)。
筛选数据:
1.关键词检索:癌症名称,研究目的,同义词等
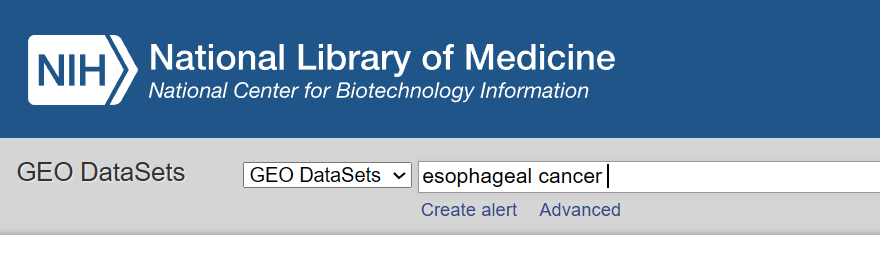
2.筛选:物种,数据类型,研究类型
【为了避免筛掉所需的数据集,一般只筛物种,对于临床研究来说,选择homo,也就是人类,更具有可信度。之后点进去查看详细信息,如果是看基因表达的,mRNA的,可以查看是不是mRNA,或者是不是transcription的。其他待补充】
-
若研究基因表达→选
Expression profiling by array。 -
若研究表观遗传→选
Methylation profiling by array。 -
若需特殊分子或整合分析→选
Customize
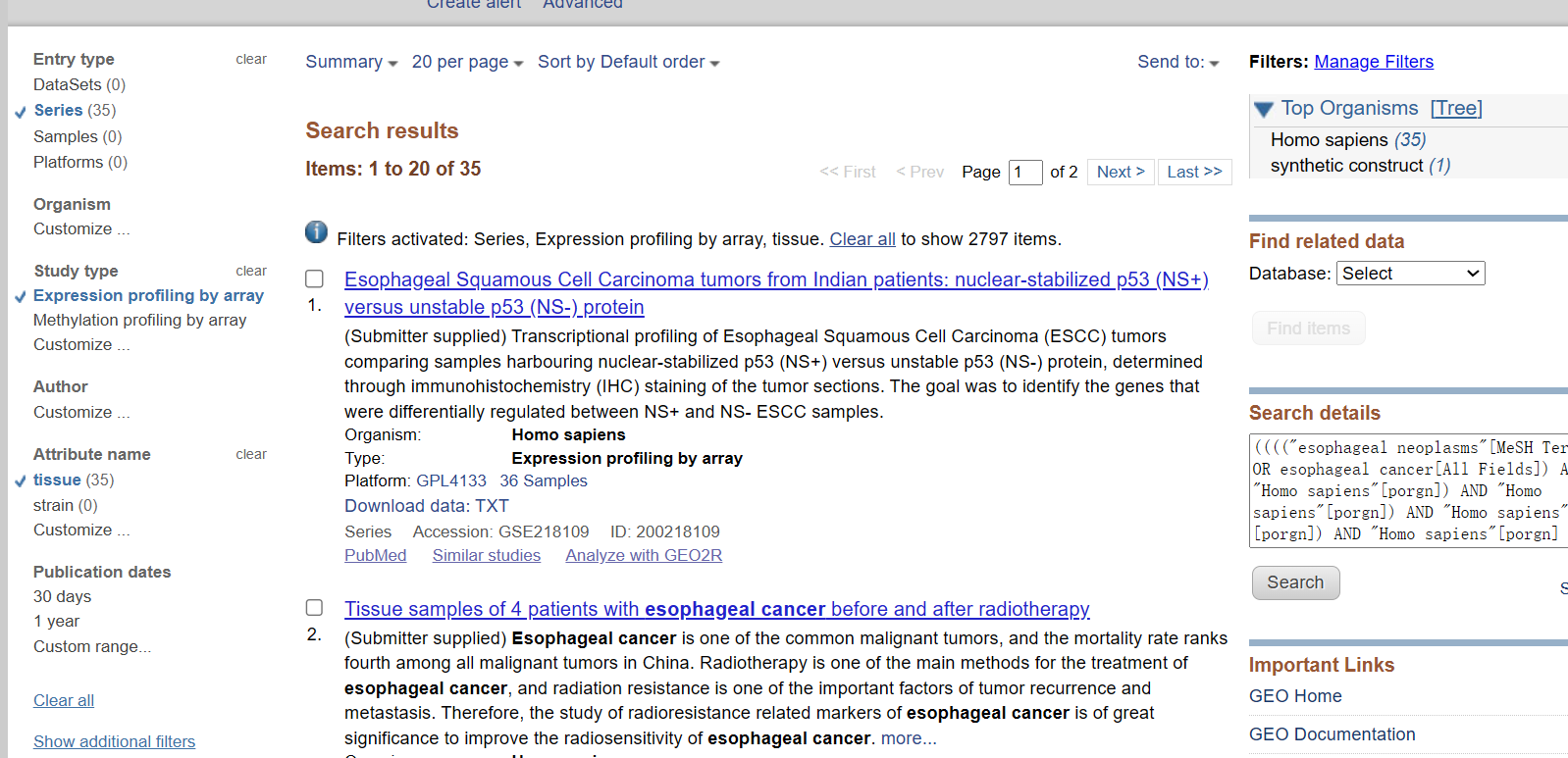
3.筛选高质量数据库:
样本量大于30;
实验设计包括癌组织和癌旁正常组织对比;
4.下载数据
选择Series Matrix File(s),这里会有三个部分信息,一是GSE的一些信息,二是临床信息,三是表达矩阵。

二、获取注释信息
获取注释信息的方式主要有三种。第1、2种一般来说都会有,不是第1种就是第2种,第3种是一些常用数据集有,不一定都有。
点进去相对应的平台文件信息后,划到最下方
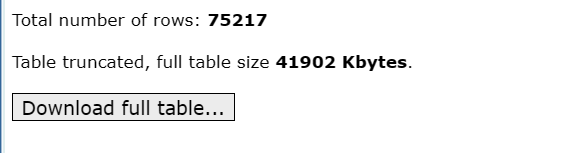
1.TXT格式
点击Download full table即可
2.Soft格式
如果出现View full table,就点击SOFT formatted family file(s)
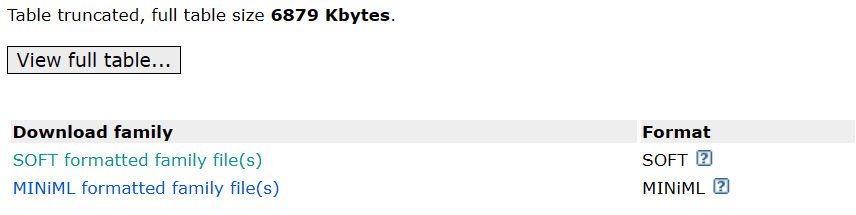
进去之后,再点击下载soft.gz格式的文件

3.对应平台的R包
以下链接是转载的,写的很详细,有链接来源,最全的R包,代码:
https://blog.csdn.net/weixin_40739969/article/details/103186027
三、注释及整理
不同方式获取的注释信息对应不同的注释及整理方式。
先解压,先解压,先解压文件!!!
跑对应的代码即可
1.TXT格式
#加载
library(GEOquery)
#设置工作目录
setwd("")
#下载矩阵,目录下已存在,自动读取
#获取表达矩阵
#如果数据已经经log后,显示log2 transform not needed;
#如果数据没有尽行log,需要log程序按照代码尽行log转换,完成后显示log2 transform finished。
#获取临床信息
#输出临床信息
#读入txt注释文件
#查看
#提取探针ID及基因symbol
#修改列名
#获取基因symbol
library(stringr)
#去掉没有注释symbol的探针
#由于部分探针ID是由全数字组成的,会导致R自动将其识别为数值型
#因此,这里需要将探针的id改为字符型
#平台文件的ID和矩阵中的ID匹配。%in%用于判断是否匹配。
#获取匹配的表达数据
#查看探针名与注释文件名是否完全一致
#合并
#删除重复基因,保留最大值
#一定注意看,是否需要删除几行
#转化行名
#删除第一二列
#导出
write.table(data.frame(ID=rownames(dat),dat),file="GSE.txt", sep="\t", quote=F, row.names = F)
2.Soft格式
#加载
library(GEOquery)
#设置工作目录
setwd("")
#下载矩阵,目录下已存在,自动读取
#获取表达矩阵
#如果数据已经经log后,显示log2 transform not needed;
#如果数据没有尽行log,需要log程序按照代码尽行log转换,完成后显示log2 transform finished
#获取临床信息
#输出临床信息
#下载soft注释文件,目录下有会自动读入
#获取注释信息
#查看
#提取探针ID及基因symbol
#修改列名
#获取基因symbol
library(stringr)
#去掉没有注释symbol的探针
#由于部分探针ID是由全数字组成的,会导致R自动将其识别为数值型
#因此,这里需要将探针的id改为字符型
#平台文件的ID和矩阵中的ID匹配。%in%用于判断是否匹配。
#获取匹配的表达数据
#查看探针名与注释文件名是否完全一致
#合并
#删除重复基因,保留最大值
#一定注意看,是否需要删除几行
#转化行名
#删除第一二列
#导出
write.table(data.frame(ID=rownames(dat),dat),file="GSE.txt", sep="\t", quote=F, row.names = F)
3.对应平台的R包
#加载
library(GEOquery)
#设置工作目录
setwd("")
#下载矩阵,目录下已存在,自动读取
#获取表达矩阵
#如果数据已经经log后,显示log2 transform not needed;
#如果数据没有尽行log,需要log程序按照代码尽行log转换,完成后显示log2 transform finished。
#获取临床信息
#输出临床信息
#提取信息
library(hgu133plus2.db)
library(dplyr)
#查看列名,获取SYMBOL
#获取探针ID及gene symbol
#修改列名
#获取基因symbol
#去掉没有注释symbol的探针
#由于部分探针ID是由全数字组成的,会导致R自动将其识别为数值型
#因此,这里需要将探针的id改为字符型
#平台文件的ID和矩阵中的ID匹配。%in%用于判断是否匹配。
#获取匹配的表达数据
#查看探针名与注释文件名是否完全一致
#合并
#删除重复基因,保留最大值
#一定注意看,是否需要删除几行
#转化行名
#删除第一二列
#导出
write.table(data.frame(ID=rownames(dat),dat),file="GSE.txt", sep="\t", quote=F, row.names = F,col.names = T)
四、导出
1.临床信息
整理:删去不需要的信息,保留所要的,比如性别、年龄、分期、生存状态等

















 2375
2375

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









