







#问题来源
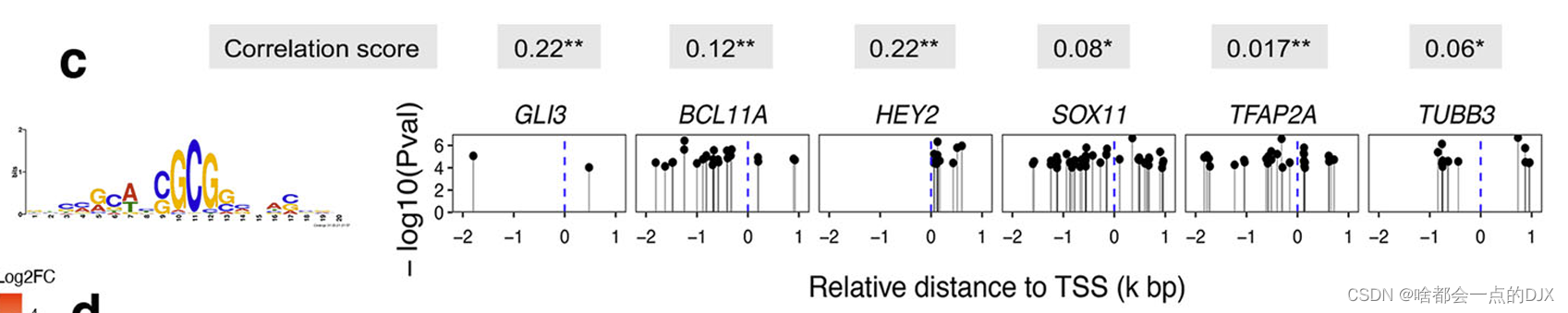
博主近期阅读一篇文献,《Transcriptomics of Hirschsprung disease patient-derived enteric neural crest cells reveals a role for oxidative phosphorylation》,作者使用FIMO meme分析了关键转录因子HDAC的motif(可视为是该转录因子的结合位点的序列特征)可能的靶基因启动子结合位点,以确定后续的研究分子。图片描述:Motifenrichment of HDAC1 at the promoter region (−2 to 1kb relative to TSS) of its target genes.
HDAC1DNA-binding motif is shown in the left panel. TSS transcription start site. P
values (Pval) were calculated by meme FIMO software.
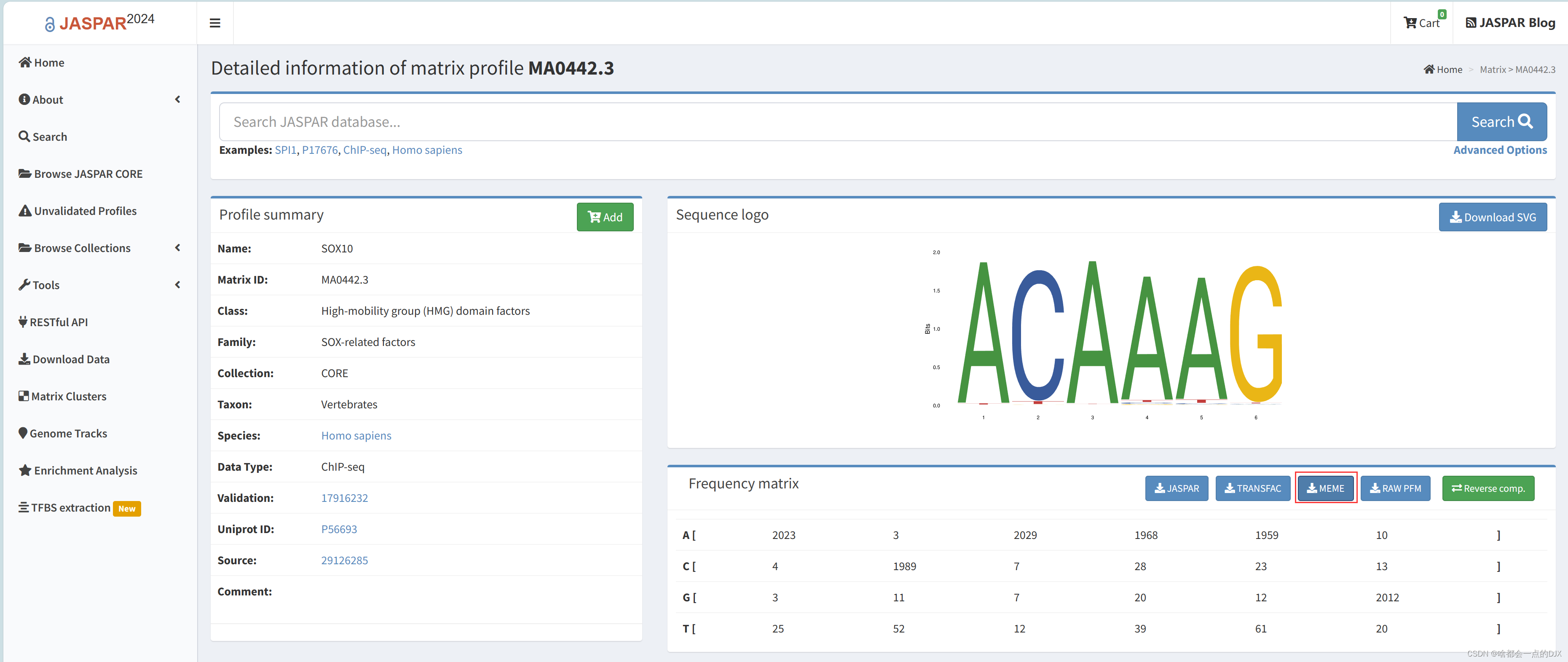
FIMO meme的使用说明参照https://www.jianshu.com/p/37eadb126bbf,需要准备1.转录因子motif,可自jaspar数据库下载(https://jaspar.elixir.no/)或自行下载序列分析,该数据库可下载ChIP-seq中相应蛋白质结合DNA序列的特征;
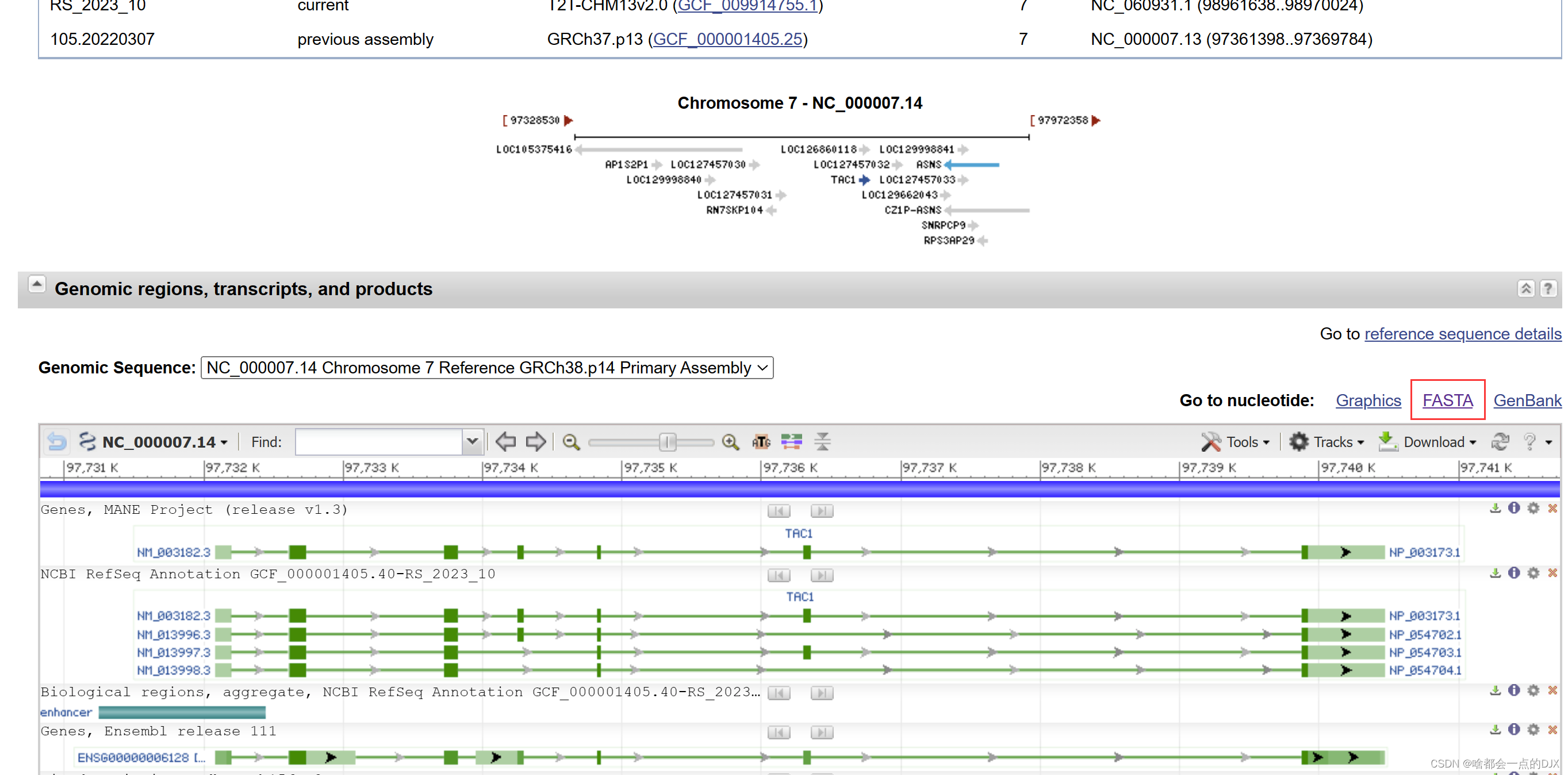
2.靶基因的启动子序列fastq(转录因子起始位点(TSS)上游2kb-下游1kb),自NCBI网站下载,注意需要手动改序列范围

但是,当做大通量筛选的时候需要一个一个复制序列文件,非常麻烦!于是博主查阅资料,发现大多数是下载全序列的,并不能指定序列的区域。于是博主写了一个自动下载的代码,主要使用Rcurl爬取静态网页和Python selenium爬取动态网页批量下载根据TSS指定范围的序列,整合后进行大批量FIMO分析。参考blog《R语言批量爬取NCBI基因注释数据》(https://www.jianshu.com/p/4236a40adeb9)、《selenium入门超详细教程——网页自动化操作》(https://blog.csdn.net/kobepaul123/article/details/128796839)。
#R环境准备
##加载包
library(RCurl)
library(stringr)
library(XML)
library(clusterProfiler)
library(httr)
library(reticulate)
##参数设置
#parmameter
#genes: a character contains gene SYMBOL
genes <- c("PHOX2B","RET","PHOX2A","NOTCH1","S100B")
#start_: sequence start position compare to TSS
start_ <- -2000
#end_: sequence end position compare to TSS
end_ <- 1000
##Rcurl配置
下载ssl证书并指定存放位置,下载地址https://curl.se/ca/cacert.pem,设施访问header(从浏览器网页,按F12进入开发者模式获取)
#set SSL certificate position
options(RCurlOptions = list(cainfo = "D:/R/cacert.pem",
ssl.verifypeer=FALSE))
myheader <- "Mozilla/5.0 (Windows NT 10.0; Win64; x64) AppleWebKit/537.36 (KHTML, like Gecko) Chrome/122.0.0.0 Safari/537.36 Edg/122.0.0.0"
##一些函数,引用自前述的blog
# get node text accroding to xpath:
getNodesTxt <- function(html_txt1,xpath_p){
els1 = getNodeSet(html_txt1, xpath_p)
# remove " ":
els1_txt <- sapply(els1,xmlValue)[!(sapply(els1,xmlValue)=="")]
# remove "\n":
str_replace_all(els1_txt,"(\\n )+","")
}
# The format of the processing node is character and the value of length 0 is NA:
dealNodeTxt <- function(NodeTxt){
ifelse(is.character(NodeTxt)==T && length(NodeTxt)!=0 , NodeTxt , NA)
}
##基因转换为Entrze ID,并生成NCBI网址
# change gene symbol to entrze ID:
genes <- bitr(genes, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
# get NCBI url
genes$NCBI_url <- paste("https://www.ncbi.nlm.nih.gov/gene/",genes$ENTREZID,sep="")
head(genes)
##使用循环下载数据,分为几个步骤
###1.使用Rcurl包进入NCBI对应基因的网站,获取Summary信息,并使用xpath定位并获取fasta的网址
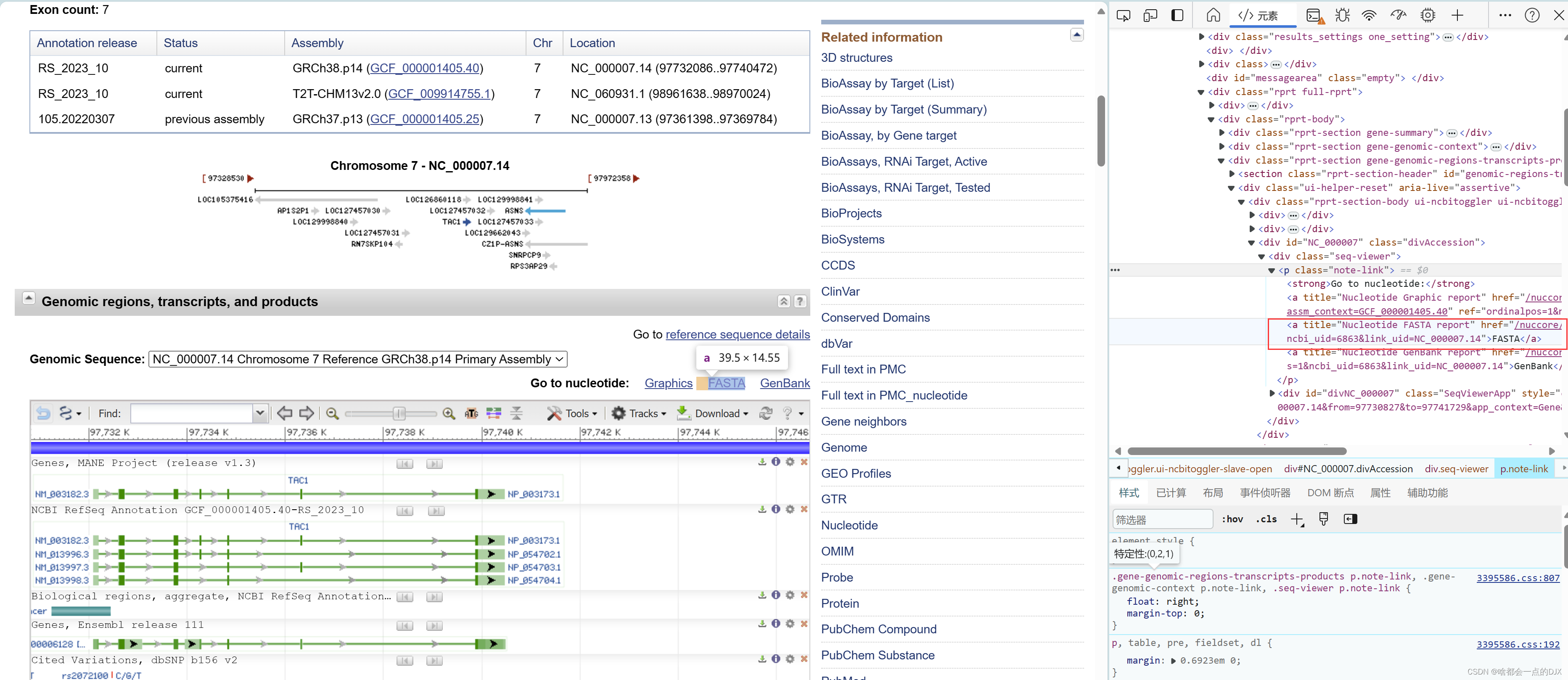
###2.编辑fasta序列网址,以https://www.ncbi.nlm.nih.gov/nuccore/NC_000007.14?report=fasta&from=97732086&to=97740472为例,该网址from参数是转录起始位点(TSS),我们需要转录起始位点前2kb-后1kb的序列,因此获取from参数与start_相加得到新的起点,与end_相加得到新的终点,随后再拼接为新的网址
###3.随后进入新的网址,这里博主最初用Rcurl爬取后发现并无序列内容,查阅资料发现NCBI的序列是动态加载的,因此需要使用处理动态网页的工具进行爬取。博主最初打算在R中跑完,但发现Rwebdrive包和Rselenium包不是很好用,配置麻烦而且对新版本的selenium兼容性不是很好,因此采用了一小段python代码进行爬取,注意检查计算机的python环境。
####cmd中运行where python查看python位置,然后[python.exe路径] -m pip install selenium
####4.根据浏览器版本下载dirver,这里用的是edge浏览器 https://developer.microsoft.com/en-us/microsoft-edge/tools/webdriver/?form=MA13LH
####5.python脚本,构建爬取函数(缺少模块的话pip安装一下),大致流程是根据提供的url开启浏览器模拟动态访问,等待5秒加载之后获取网页内容,在定位序列位置
#python script
import time
import lxml.html as lh
from selenium import webdriver
from selenium.webdriver.chrome.service import Service
def getseq(url):
options = webdriver.EdgeOptions()
edge_path="D:/R/msedgedriver.exe" #edge driver path
service = Service(executable_path=edge_path)
options.add_argument('--headless') #not open browser window
browser = webdriver.Edge(options=options,service=service)
browser.get(url)
time.sleep(5) #wait for page load
xpath = '//*[@id="viewercontent1"]/pre/text()'
html_source = browser.page_source
html = (lh.fromstring(html_source))
items = html.xpath(xpath)
browser.close()
return items
####6.在R中加载python脚本,这样就有构建的函数了,并且可以直接在R中调用python
source_python("D:/Python/pythonProject/NCBI-DNA-sequence/Main_script.py")
####7.在R中调用getseq函数获取序列及基因表达位置信息,整合到genes中
循环完整代码如下:
for(i in 1:nrow(genes)){
# get gene web:
doc <- getURL(genes[i,"NCBI_url"],header=myheader)
html_txt1 = htmlParse(doc, asText = TRUE)
# get summary:
genes[i,"Summary"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[preceding-sibling::dt[text()="Summary" and position()=1 ] ]'))
cat("write Summary\n")
# set sequence web
url_seq <- unlist(getNodeSet(html_txt1,"//a[@title=\"Nucleotide FASTA report\"]/@href"))
url_seq <- paste0("https://www.ncbi.nlm.nih.gov",url_seq)
url_split <- strsplit(url_seq,"=")
TSS_point <- url_split[[1]][3]
TSS_point <- gsub("&to","",TSS_point)
if (exists("start_")){
start_point <- as.numeric(TSS_point)+start_
genes[i,"Start"] <- start_point
url_split[[1]][3] <- paste0(start_point,"&to")
url_seq <- paste(url_split[[1]],collapse = "=")
}else{
genes[i,"Start"] <- TSS_point
}
if (exists("end_")){
end_point <- as.numeric(TSS_point)+end_
genes[i,"End"] <- start_point
url_split[[1]][4] <- end_point
url_seq <- paste(url_split[[1]],collapse = "=")
}else{
genes[i,"End"] <- url_split[[1]][4]
}
print(url_seq)
# get sequence web
doc <- getURL(url_seq,header=myheader)
html_txt2 = htmlParse(doc, asText = TRUE)
gene <- getNodeSet(html_txt2,"//*[@id=\"viewercontent1\"]/pre/text()")
#get sequence
seq <- getseq(url_seq)
seq_split <- strsplit(seq," ")
genes[i,"Ano"] <- gsub(">","",seq_split[[1]][1])
genes[i,"chromosome"] <- gsub(",","",paste(seq_split[[1]][2:5],collapse = " "))
genes[i,"Ref"] <- seq_split[[1]][6]
genes[i,"Seq"] <- gsub("Assembly","",seq_split[[1]][8])
cat("write sequence\t")
print(paste("complete ",i,"genes!"))
}
###生成序列整合文本
#intergrate seq file
seq_inter <- paste(paste0(paste0(">",genes$SYMBOL),genes$Seq),collapse = "")
write.table(seq_inter,"genes_seq_intergrate.txt",col.names = F,row.names = F,quote = F)

这样就可以直接上传到FIMO中进行scan了!
#小结
这篇教程中综合了Rcurl和Python selenium的方法,对动态网页的数据进行爬取和整合,可以任意指定基因序列进行下载。然而爬取的速度较慢,仍有优化的部分,如对每个url都调用了getseq方法,导致每个基因都会打开并关闭一次浏览器,增加了任务时间等。
















 7049
7049











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











