







论文笔记_大作业PPT: 这是我自己做的配套PPT,仅供参考 (gitee.com)
今天我给大家介绍的文章是《Accurate structure prediction of biomolecular interactions with AlphaFold 3》。也就是今年斩获诺贝尔化学奖的AlphaFold2的升级版,研究领域是生物分子结构预测,而不仅仅是蛋白质结构预测。于今年5月8日在Nature发表,当时是没有开源的,上个月AF3开源了,所以想和大家分享这篇论文。

先和大家分享一个很有趣的故事,在2021年7月15日,Google的DeepMind团队发表AlphaFold2,并占领了Nature期刊8月份的封面。而就在同一天,也是7月15日,华盛顿大学的Protein Design团队发表了RoseTTAFold,并占领了Science期刊8月份的封面。也就是说,两篇论文几乎是在同一时间发表,而且都是封面文章。我在阅读AlphaFold3时还不知道RoseTTAFold,但文章中多次与这个模型进行对比,基本上把它当作AlphaFold3最大的对手。于是我进一步查找这个模型的资料,才知道有这么一个故事,不知道两个团队在发论文前有没有提前商量好。
一、背景
自然界每一种植物、动物和人类细胞内部,都包含有数以亿计的分子机器。这些分子机器由蛋白质、DNA、RNA及其他配体分子组成。正是这些由生物分子组成的小型机器,维持着生命的运转和延续。DNA 将遗传信息传递给 RNA,再翻译成蛋白质,进一步折叠成二级结构、三级结构、四级结构。大多数蛋白质会折叠成独特的构象,而结构所需的信息都编码在氨基酸序列中,也就是我们常说的:序列决定结构,结构决定功能,蛋白质结构预测对于了解生物功能至关重要。就像钉子和螺丝刀,都是铁做的,但因为两者的结构不同,所拥有的功能便大相径庭。
三年前,AlphaFold 2 在蛋白质结构预测方面大幅领先其他算法,而现在,AlphaFold3不仅能够预测蛋白质结构,还能对核酸、小分子、离子以及化学修饰等和蛋白组成的复合物进行结构预测,且准确性有巨幅提升。这表明,通过单一深度学习框架,也许真的可以预测所有生物分子相互作用的高精确度结构。举个例子,7PNM是一种普通感冒病毒的刺突蛋白,AlphaFold 3预测的结构与Ground truth对比。其中刺突蛋白是蓝色的、抗体是青绿色,简单糖是黄色的,Ground truth是灰色的。像这样的预测,AlphaFold 3做了很多,有助于我们更好地理解冠状病毒,包括COVID-19,为改进治疗提供可能。
二、模型架构
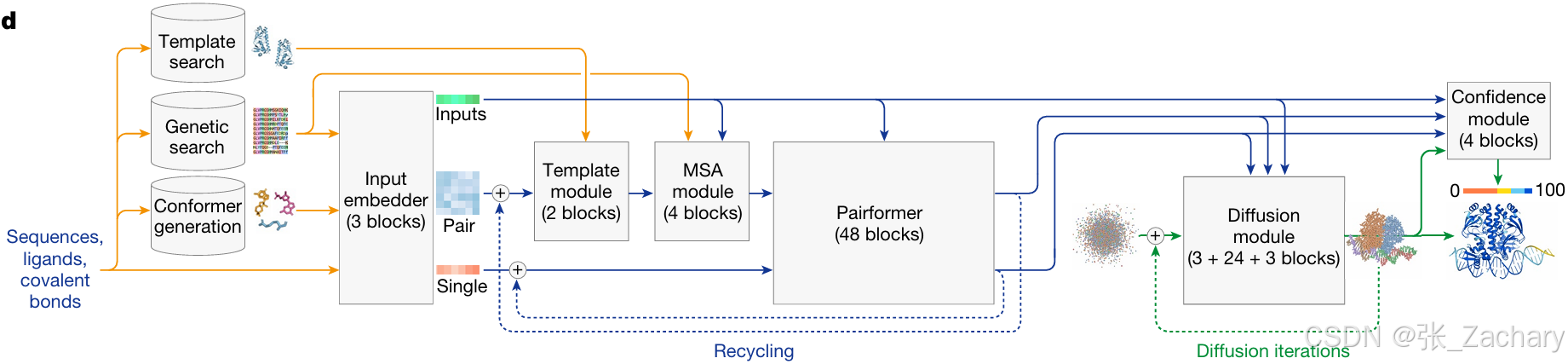
AF3模型大致可分为四个部分。
2.1输入模块
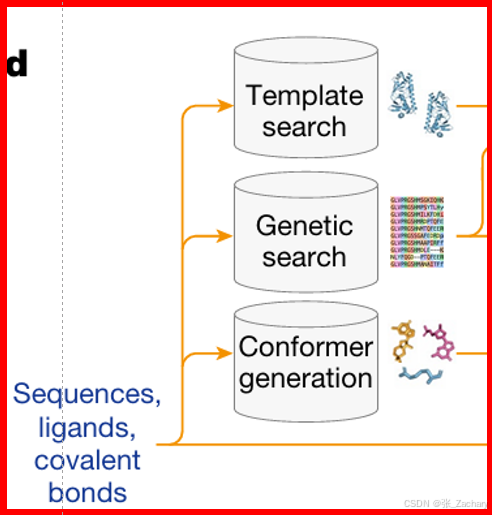
向模型输入序列信息,如图示。之后模型自动执行以下三个操作:
1、从 PDB mmCIF 文件中检索模板,选择搜索到的前4个模板输入到下一部分。模板具有与目标蛋白质相似的结构和功能,可以作为参考来构建目标蛋白质的模型。需要注意的是模板搜索仅针对单链的蛋白质进行,不提供多链的模板信息。
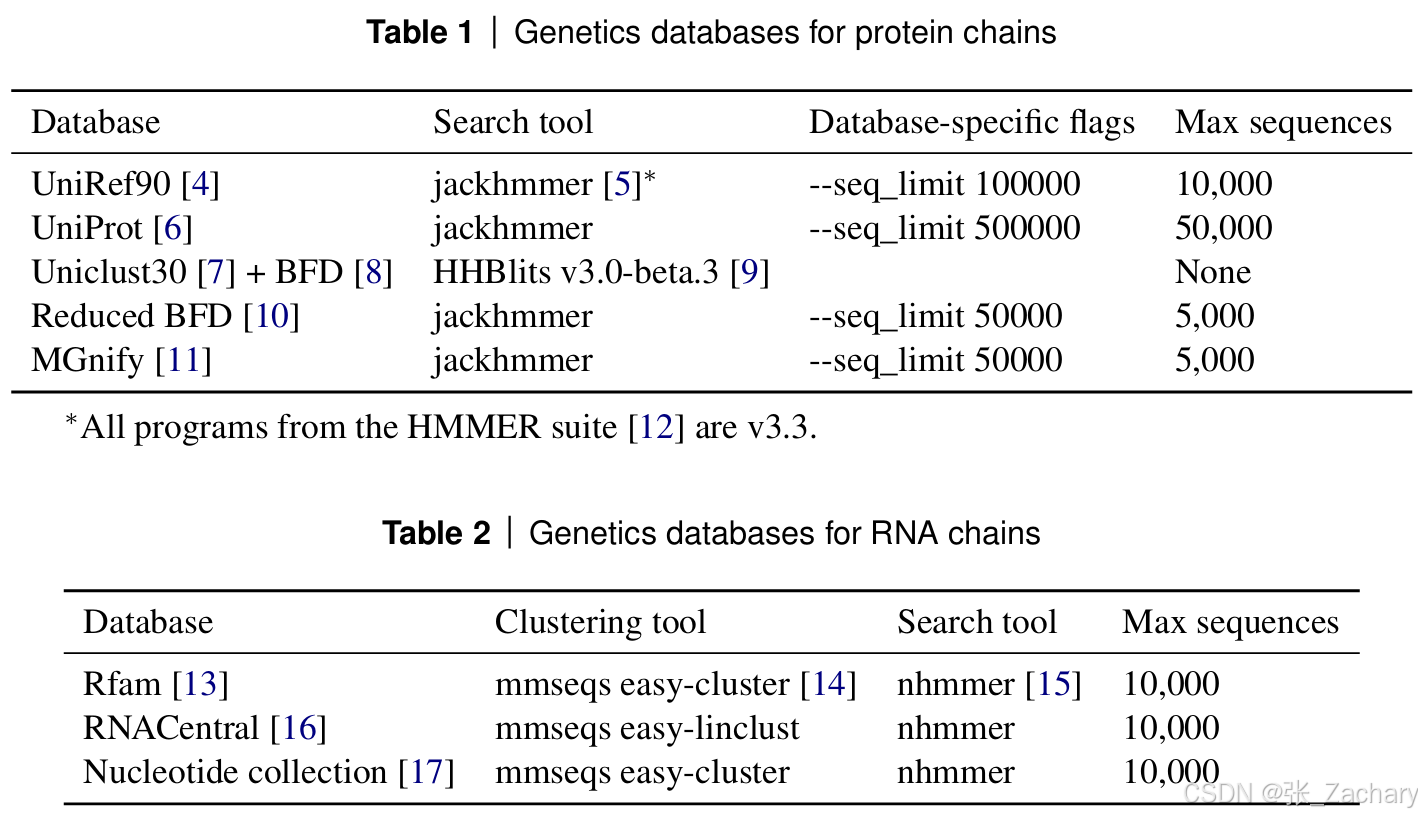
2、从 5 个数据库中检索蛋白质序列,从 3 个数据库中检索 RNA 序列,用于多序列比对(MSA),如下图所示。
3、生成参考构象,也就是氨基酸、核苷酸或配体应该长什么样子。给定输入的CCD代码(一种用于标识化学组分的简短字符串编码系统)或SMILES字符(一种用来描述分子结构的字符串表示方法),使用RDKit(一种开源的分子信息学工具包)的ETKDGv3方法(一种分子构象生成算法)生成构象。
第二步检索到的相似序列、第三步生成的构象、以及原始的输入信息,将会输入到特征提取模块。第一步的模板信息token化后,会直接进入编码器的Template module。
2.2特征提取模块
先将输入进来的信息token化,token就是将生物语言用数字表示,比如用1~20表示氨基酸。文章要求:每个氨基酸残基对应于一个token;每个核苷酸残基对应于一个token;对其他分子,将每个重原子记为一个token。比如,35个标准氨基酸的序列(可能大于600个原子)将由35个token来表示;同时,一个由35个原子组成的配体也同样由35个token表示。
再为token的每一个属性赋值,每一个token最终都会由一个向量来表示。比如半胱氨酸这个氨基酸,氢键供体属性赋值为 0.5,氢键受体属性赋值为 0.6,具有可旋转化学键属性赋值为 0.3,表面电荷属性赋值为 -0.88......可以选择许多属性来描述token。Alphafold3为每个token 选择了384个属性,故每个token会被表示成一个长为384的向量。之后进行embedding操作,将信息映射到向量。
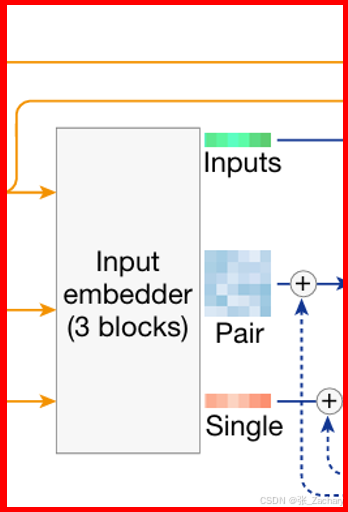
Input embedding:每条输入序列会被表示成一个(n x 384)的向量,其中n表示该序列中包含token的数量。从模型数据流可以看出,Input embedding后得到的representation不参与后面的循环更新,但是它参与后面每一个模块的计算。
Single embedding:它也是一个(n x 384)的向量,是由 Input向量乘一个权重矩阵而得出。这个权重矩阵是可学习的,权重矩阵会通过反向传播算法在训练中不断优化,如果熟悉Attention对这个应该不陌生。因为Input representation和single representation并不关注token在序列中的顺序与位置,所以还需要一个pair representation,来利用关于位置等和两两序列之间相关的信息。
Pair embedding:它表示两两序列之间的关系,这两个序列是同一类型的,比如氨基酸序列与氨基酸序列。所以蓝色方阵Pair的维度为(n x n x 128),n表示该种序列中包含token的数量,若两个序列token数量不一,会先进行对齐操作。128指的是两两序列之间有128种信息可以学习,比如两个序列的相对位置信息,化学键信息等。
2.3编码器
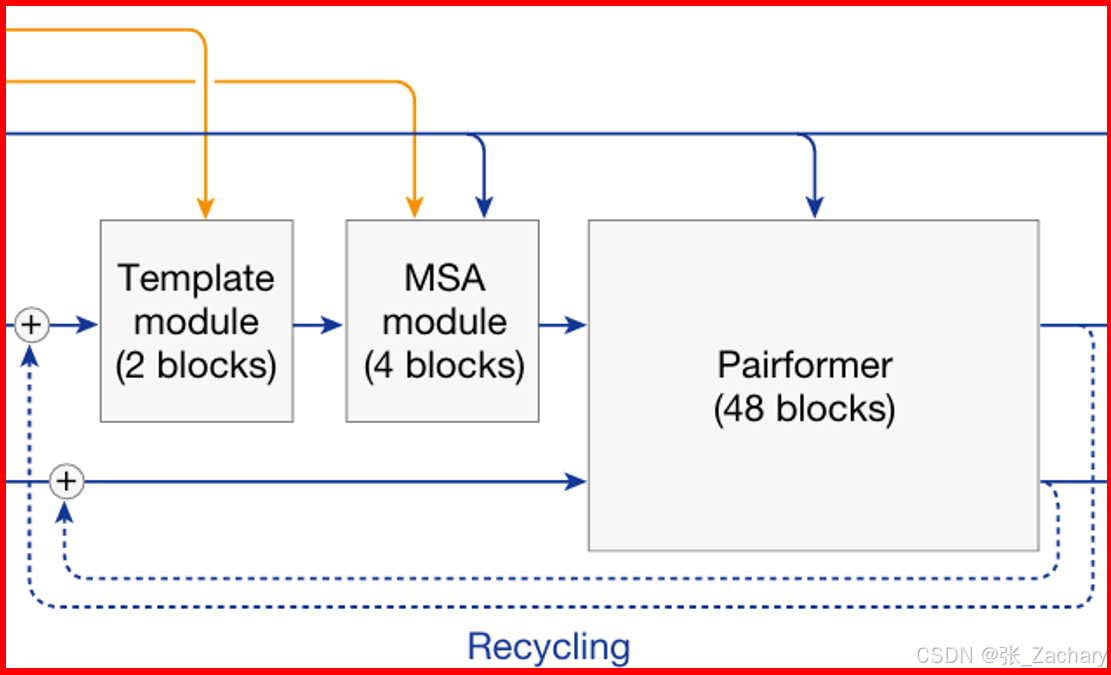 Template module:在结构数据库中检索到的模板信息会在这里与Pair embeddings整合,着重关注对结构更重要的区域,最后得到Pair representation。
Template module:在结构数据库中检索到的模板信息会在这里与Pair embeddings整合,着重关注对结构更重要的区域,最后得到Pair representation。
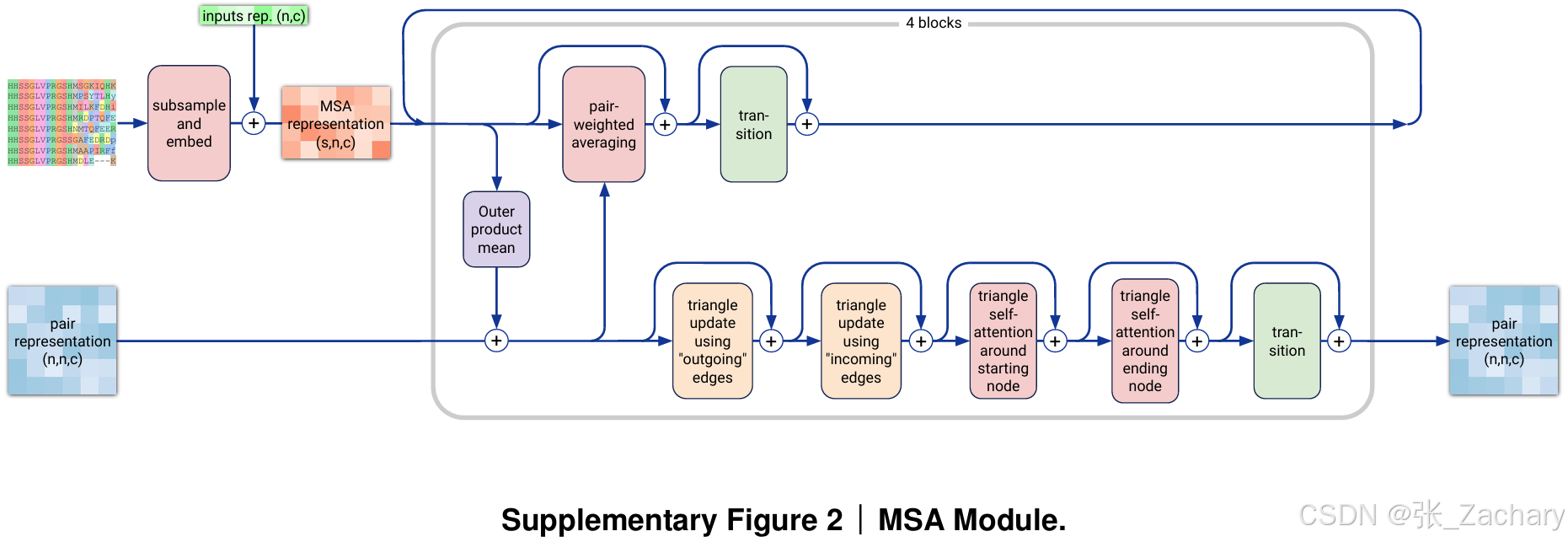
MSA module:添加多序列比对信息,更新 Pair representation。MSA模块通过随机抽取序列和embedding操作为每个Input representation生成MSA representation。多序列比对的目的是看一看数据库里有没有和我们输入的序列相似的序列,以便得知同一个token在不同序列的表现,比如同一个氨基酸在不同蛋白质的表现。MSA representation的维度为(s x n x 64),s表示从数据库中抽取到多少条序列,比如鱼的某个蛋白质序列,兔子的某个蛋白质序列,猩猩的某个蛋白质序列,再加上我们输入的序列;n是每条序列中token的数量,比如蛋白质序列中的氨基酸个数;64是一个超参数,用来表示一个token的信息,不同于之前的384,这里的64没有实际意义,是一个经过调优的平衡值,既能保证模型在学习多序列比对信息时有足够的容量,又不会导致过高的计算和内存需求。
该模块包括四个均匀的块,这些块反复处理和组合Pair representation和MSA representation。每一行MSA的注意力是独立执行的,而不再是通过传统的键-查询(key-query)机制。所有的注意力权重完全来自Pair representation,而没有跨行的直接信息融合。这种做法减少了计算和内存消耗,并且让每一行的MSA都能通过相同方式进行信息融合,简化了模型的计算。这一设计的核心思想是Pair representation承担了更多的信息融合任务,确保了蛋白质或核酸的详细信息能充分传递到网络的其他部分。
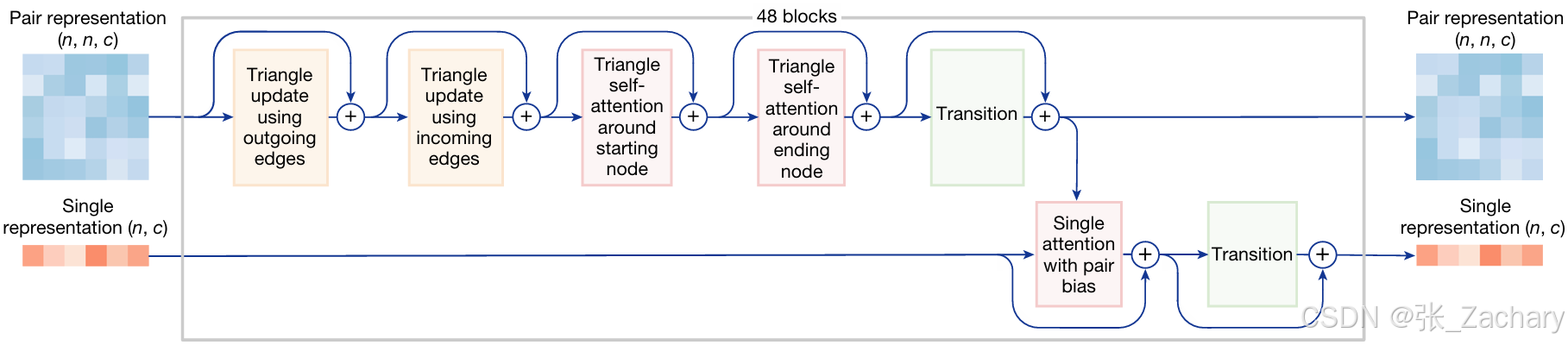
Pairformer module:是一个 Transformer module,可以很好地整合信息与聚焦关键信息,用于更新 Pair representation 和 Single representation。四次输入-输出循环后将Pair representation和Single representation 输出到解码器,这个循环过程也叫自蒸馏。这一模块与MSA基本是一致的,不同的地方在于只有Pair的信息流向Single,而Single的信息不会流向Pair,以及Single representation 更新时需要用到的Single attention with pair bias块。
2.4解码器
Diffusion Module:使用扩散模型预测复合物结构,具体做法就是,Single representation 信息和 Pair representation 信息会转变为空间位置,这些信息刚开始看上去还是杂乱无章的原子点,看不出结构信息,但是经过扩散模型的多次(加噪和去噪)更新之后,杂乱无章的原子点变成了清晰的、有意义的三维复合物结构。
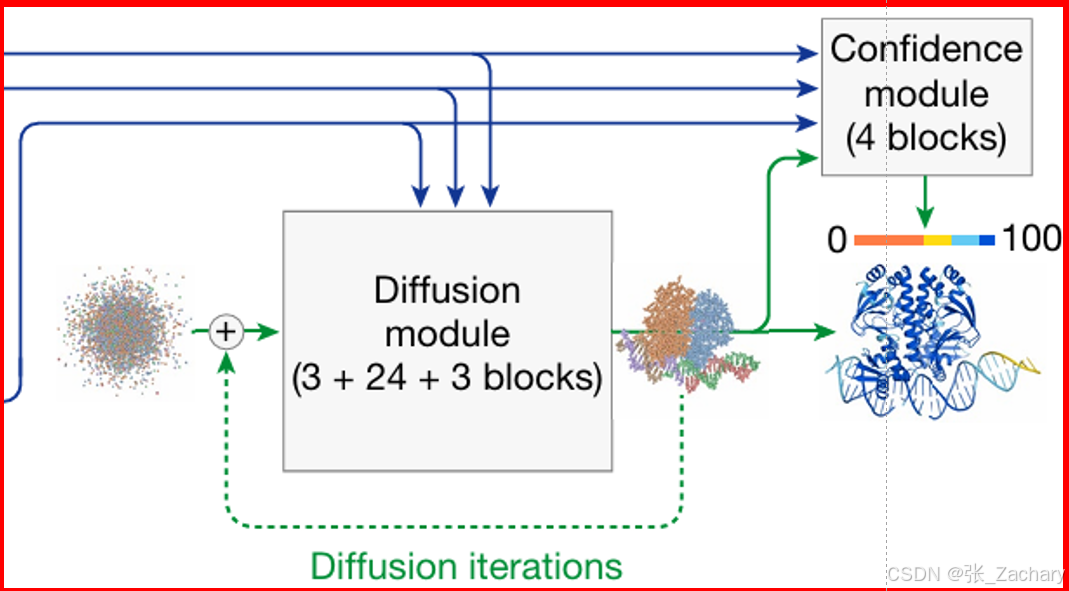
输入有三种,分别是token级别的表征,原子级别的表征和原子的真实物理坐标。token级别的表征会进入全局注意力模块,原子级别的表征会进入局部注意力模块,真实物理坐标进行随机旋转和平移,即为原子坐标添加随机的高斯噪声,有大噪声与小噪声,大噪声是针对宏观结构的,可以理解为整个蛋白质或整个多分子复合物的坐标受到微小扰动,小噪声针对局部精细结构,例如原子之间的短程相互作用和几何形状,可以理解为每个原子坐标都受到独特的微小扰动,某些原子的坐标加了一个微小值,某些原子的坐标减去一个微小值。多次加噪后,整个结构就很混乱了,模型需要学会从高噪声状态恢复到原子级的精确结构,即逐次更新原子的坐标,直到恢复与最初坐标非常接近的状态,甚至充分发挥了扩散模型的创造力,比最初坐标还能更好描述这个大分子。坐标之后会依次经过局部注意力模块、全局注意力模块、局部注意力模块。最后去噪,得到输出。
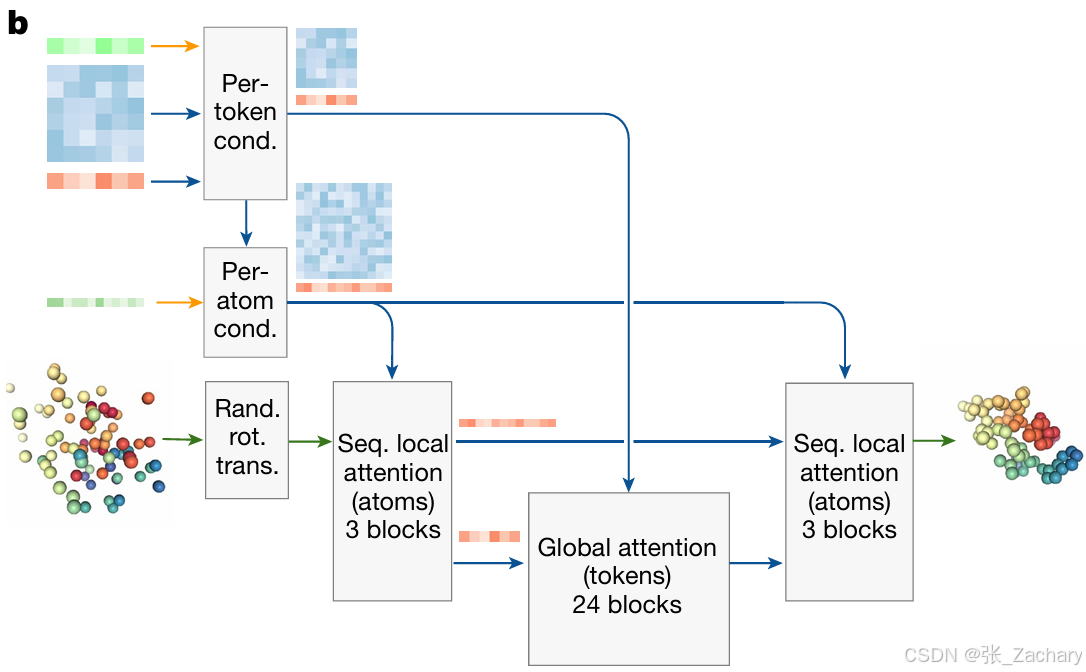
Confidence Module:作为一个裁判,对扩散模型的输出进行评价。使用预测局部距离差异检验(pLDDT,Predicted local distance difference test )、预测比对误差(PAE,Predicted aligned error)和预测距离误差 (PDE,Predicted distance error)来评估置信度,置信度越高,代表预测越准确,与Ground Truth越相似。主要采用的还是pLDDT(0~100),这个指标在我的AF3运行截图中也可以看到。
三、实验结果
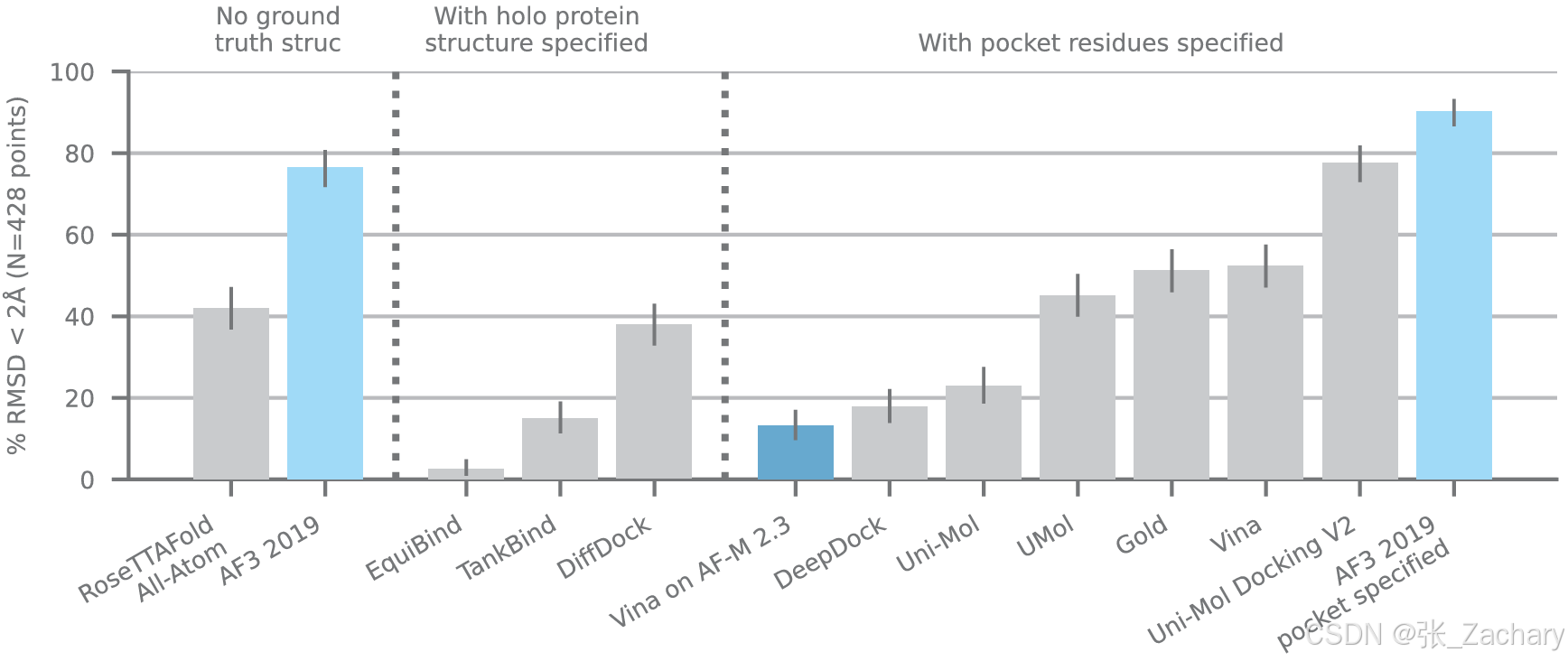
3.1蛋白-小分子
在PoseBuster Benchmark Set进行测试。这是一个用于评估蛋白质-配体对接方法性能的测试集。它包含一系列精心挑选的、公开可用的蛋白-小分子结构,并且只包含2021年1月1日到2023年5月30日之间发布的,共428个。评价指标是将预测结构和真实结构的蛋白质口袋对齐后,比较小分子之间的RMSD(均方根距离)。Baseline有两种,分别为盲对接方法和传统对接方法。在盲对接方法中,只输入蛋白质序列和小分子的SMILES码;在传统对接方法中,需要额外的结构信息,例如蛋白质的结构、指定蛋白质对接的口袋等。AF3通过盲对接方法,性能就已经超过绝大部分方法(76.4%);如果能接受结构信息,其表现更是如虎添翼(90.2%)。
3.2蛋白-核酸
Baseline是RoseTTAFold2,测试集是PDB。AF3在蛋白-RNA、蛋白-DNA、RNA-only和DNA-only的结构预测上都超过RoseTTAFold2NA。由于RoseTTAFold2NA仅针对1000个残基以下的结构进行验证,所以作者就使用最近的PDB评估集中1000个残基以下的结构进行比较。
作者还评估了AF3在10个公开可用的CASP15 RNA目标上的表现,其平均性能高于RoseTTAFold2NA和CASP15当时最好的方法AIchemy_RNA,但没有超过AIchemy_RNA2,不过该方法是基于人类专家知识辅助的,自然比纯AI要精确一些。
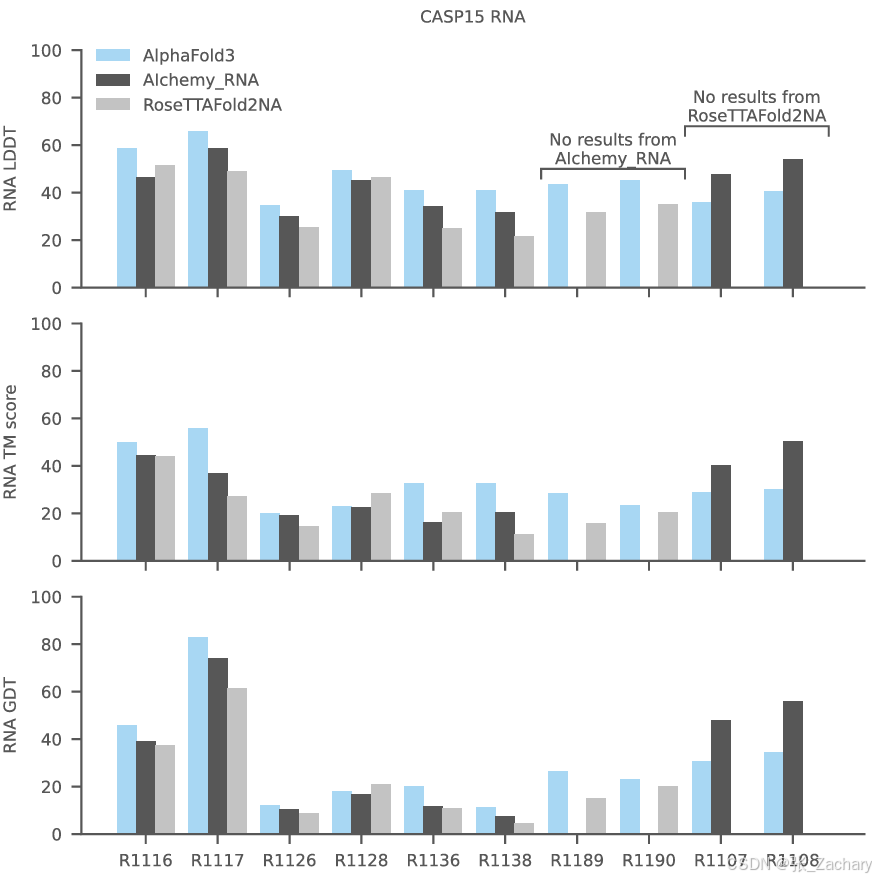
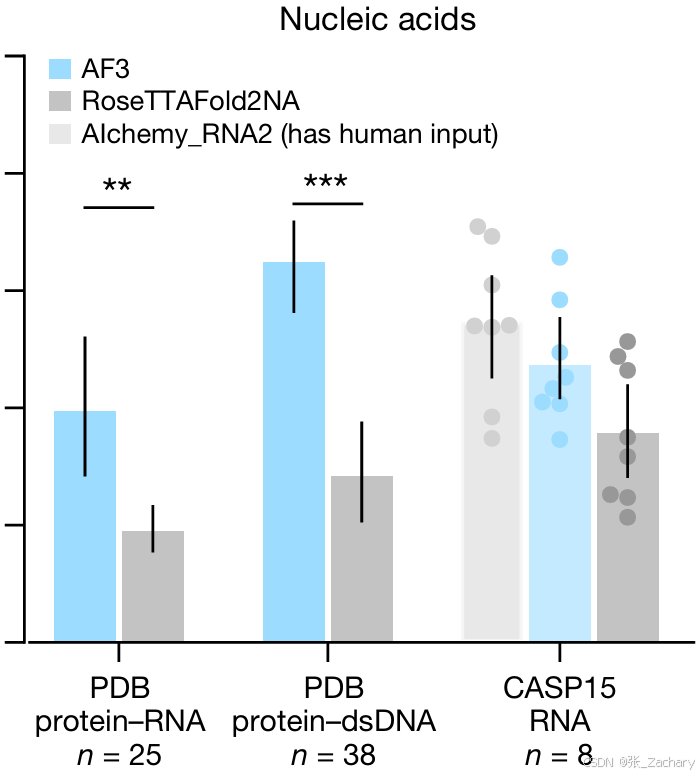
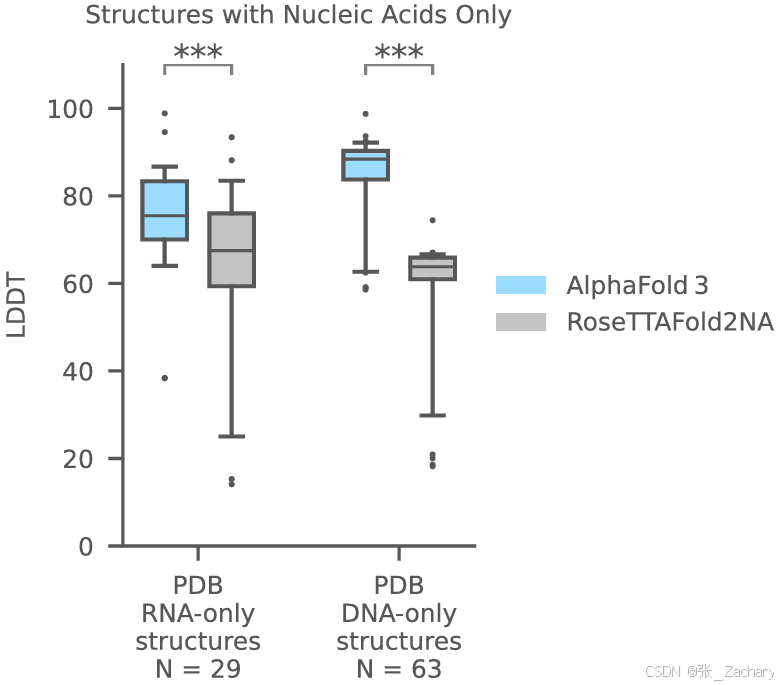
3.3共价修饰
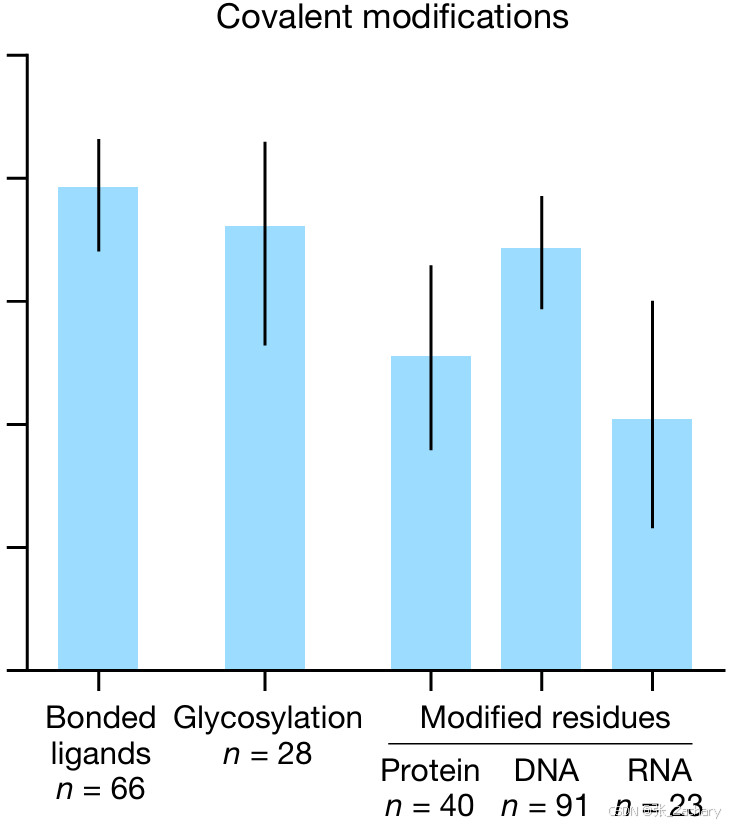
共价修饰是通过共价键将小分子或其他化学基团添加到生物大分子(如蛋白质、核酸、脂质等)上的过程。这种修饰可以改变大分子的性质、功能或稳定性,并在生物学中发挥着重要的调控作用。比如磷酸化——蛋白质上的氨基酸残基(通常是丝氨酸、苏氨酸或酪氨酸)可以被磷酸基团修饰,糖基化——蛋白质或脂质上添加糖分子,乙酰化——在赖氨酸残基上添加乙酰基团,可以影响染色质的结构和基因的表达。共价修饰也能够通过AF3准确预测,文章中给的实验结果是共价键结合配体、糖基化和修饰氨基酸、核酸残基这三种情况。
3.4蛋白质结构预测
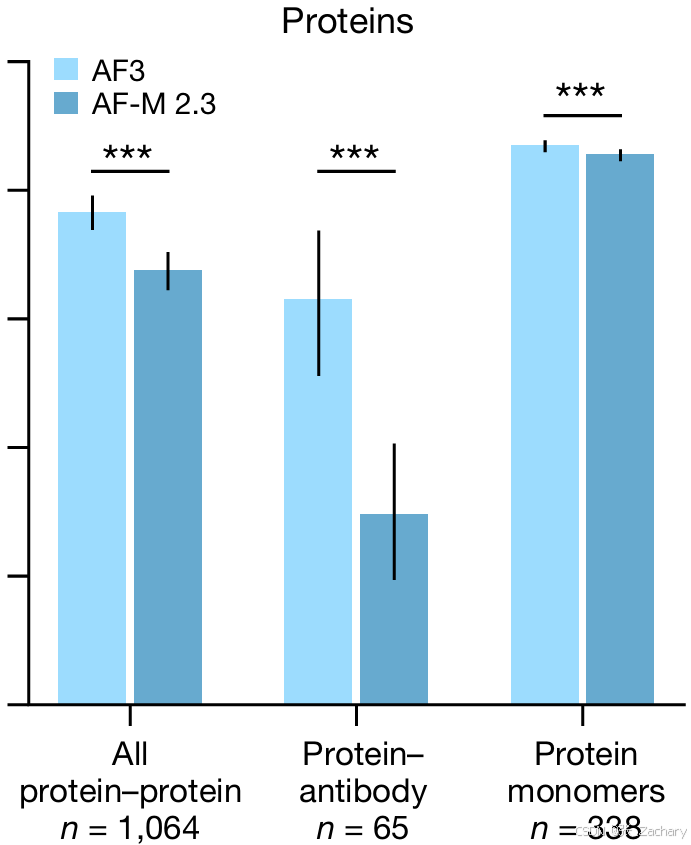
Baseline是AlphaFold-Multimer,也就是自家的上一代模型。由于 Alphafold-Mutimer 本身已经足够优秀,留下的提升空间不多,所以在预测蛋白-蛋白复合物和蛋白单体之间的结构上的能力提升不大;但在预测蛋白-抗体复合物结构的能力大幅提高。
3.5 Confidence Module置信度预测
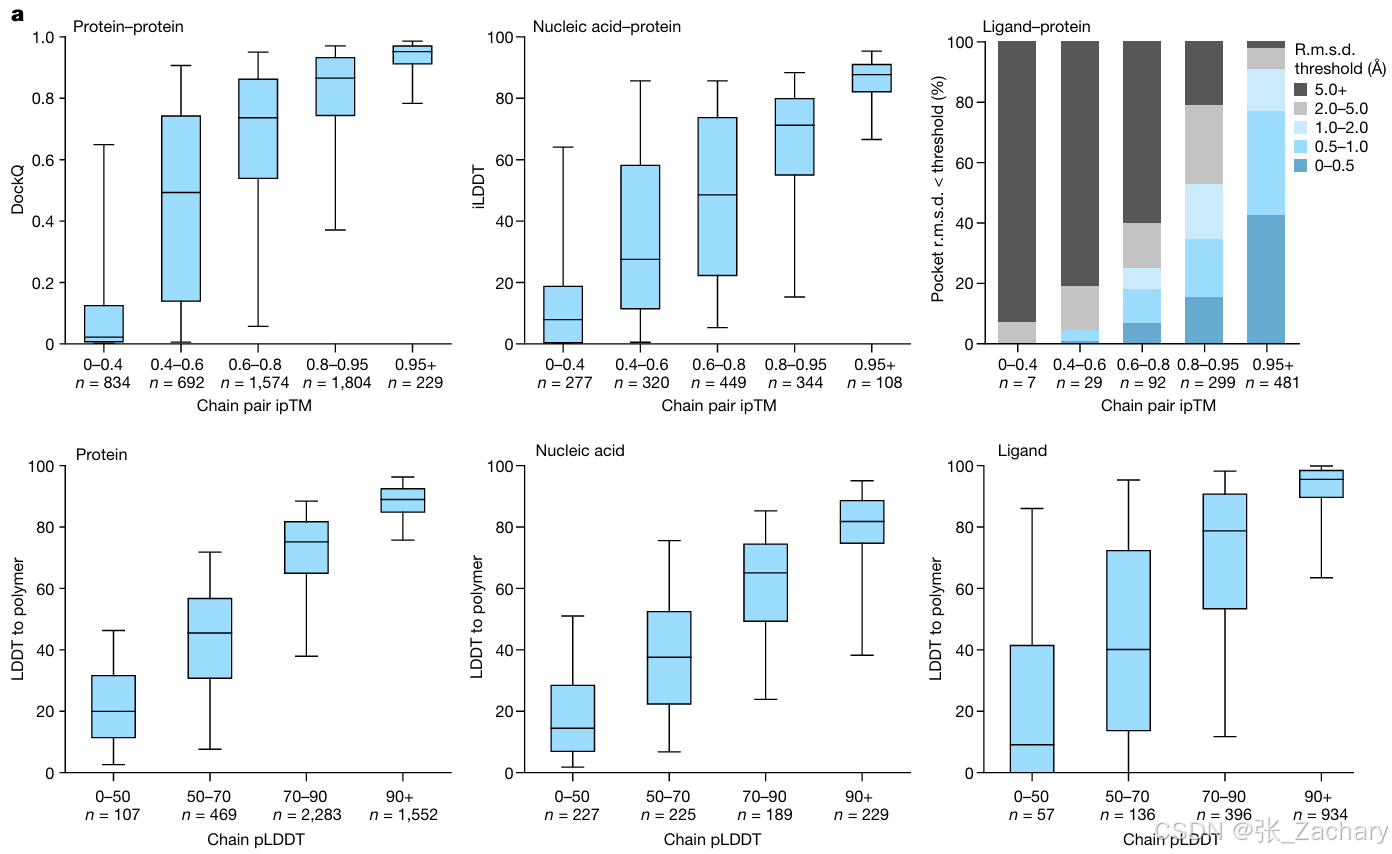
作者根据Confidence Module做了相关实验,目的是证明预测结构的准确程度与Confidence Module的置信度有很强的相关性,即Confidence Module的指标能够很好地评价模型预测的结果,而不是胡诌乱造。第一行的横坐标是作者使用的ipTM分数,是根据PAE算出来的,软件里用的也是这个指标,纵坐标是DockQ分数,这个是大家公认的很早就有的指标。第二行的横坐标是作者使用的pLDDT分数,纵坐标是LDDT分数,这个也是大家公认的很早就有的指标。实验结果显示,作者使用的指标与公认的指标之间存在显著的正相关性,也就是说作者使用的指标可以完美替代公认的指标。
四、局限性
4.1手性
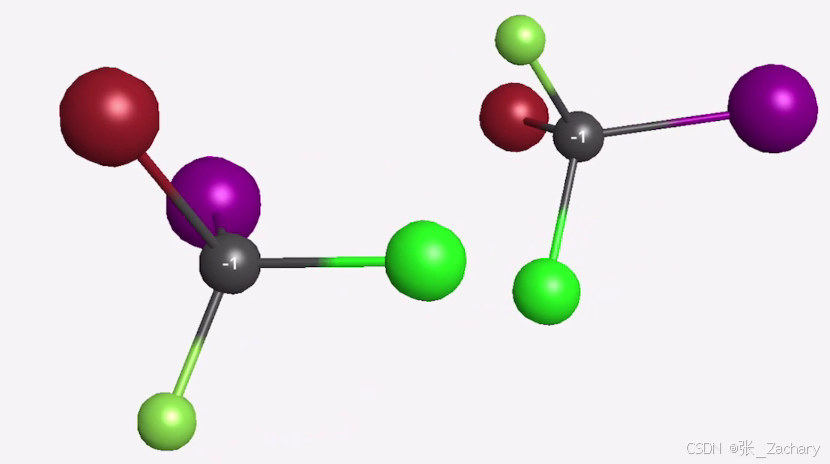
手性分子指的是两个分子不能通过平移或旋转等对称操作使物体与其镜像相重合的特性,这意味着互为手性的两个分子可能具有完全不同的性质,如图所示。左旋螺丝钉和右旋螺丝钉也可以看作“互为手性”。即使输入了正确的手性参考结构,AlphaFold3有时仍会输出违反手性的模型。尽管模型在PoseBusters基准测试中加入了手性违反的惩罚损失,但手性违反率仍有4.4%。原子重叠:在某些情况下,如蛋白-核酸复合物,模型可能会产生原子重叠的现象。尽管对这种重叠进行了惩罚以减轻问题,但这一现象并未完全消除。
4.2幻觉
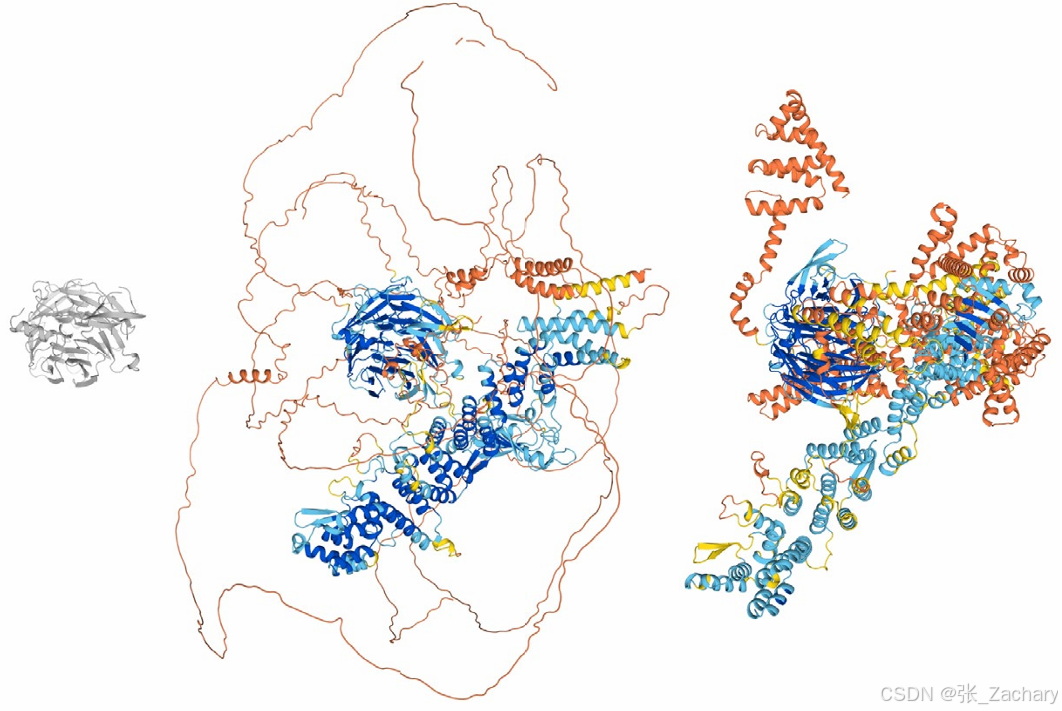
另外,AlphaFold 3 还有一个生成模型普遍存在的问题,即幻觉 (Hallucination)。 如下图的蛋白质结构预测结果,左侧的蛋白质结构仅灰色区域可解析,其余部分因电子密度不足,可能处于未折叠状态。中间的图是 AlphaFold 2 预测该蛋白质的结果,蓝色区域被认为是折叠状态,其他「彩带」部分被认为未折叠,预测的结构相对合理。右侧是 AlphaFold 3 的预测结果,它倾向于将所有可能折叠的区域均进行折叠处理,该结构看似合理,但从实际情况出发,上述大部分区域其实并未折叠。故AlphaFold 3 的幻觉倾向于将蛋白质预测为折叠状态,而非保留其可能存在的未折叠状态。
为应对 AlphaFold 3 的幻觉问题, 研究人员选择了一个直接而有效的方法:既然 AlphaFold 2 预测的结果相对合理,那就将 AlphaFold 2 预测的结果纳入 AlphaFold 3 的训练数据集中,以增强模型的训练效果。然而,该方法存在一个局限:若 AlphaFold 2 的预测本身存在错误,则可能影响到 AlphaFold 3 的预测质量,除非能引入其他的数据源来进一步优化模型。
4.3其他问题
动力学问题,AlphaFold3主要预测静态结构,而无法捕捉生物分子系统在溶液中的动态行为。这一局限性在模型中仍然存在,即使是通过扩散或使用多个随机种子,也难以获得近似的解集合。
特定目标的精度问题。在某些情况下,如E3泛素连接酶,AlphaFold3可能无法准确模拟其在不同状态下的构象。例如,它可能只预测出与配体结合时的封闭状态,而忽略了在载脂蛋白状态下的开放构象。
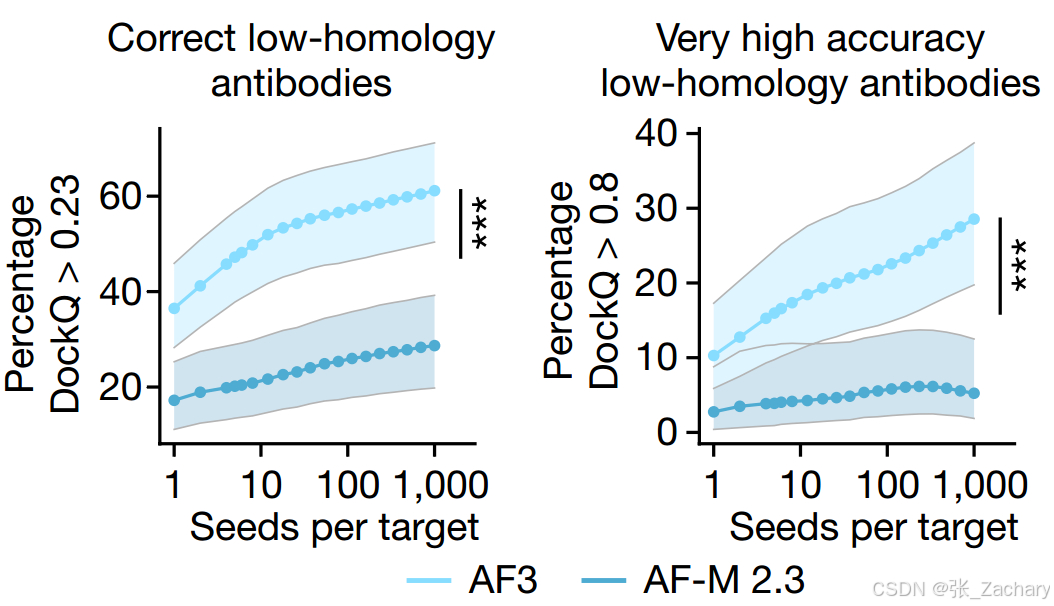
准确性与计算成本。为了提高预测的准确性,可能需要生成和评估大量的模型结果,这将导致显著增加的计算成本。特别是在抗体-抗原复合物的预测中,随着模型随机种子数量的增加,预测效果有所提升,但这也意味着需要更多的计算资源,如图所示。把每一次随机种子的选择视为一次全新的测试,当尝试的种子越多,效果就会越好。在低置信度(DockQ>0.23)的情况下,第一次尝试就能做到接近40%的成功率,提高置信度(DockQ>0.8)后,AF3的表现也还可以。这
也说明对于抗体-抗原的结合预测,还有较大提升空间。
五、我的感受
最后说一下我阅读这篇论文的感受,首先就是文章工作量特别大,我是AF2和AF3一起对比阅读,花了很多时间,可以深切地感受到Deepmind团队真的很用心,虽然很多方法是之前别人提过的,但把这些方法不断尝试组装、不断扩展模型的性能,就是一件很令人佩服的事情,这个过程需要耗费大量的人力物力财力。其次就是像这样的顶级团队没有选择缝一点模块就发一篇文章,而是把几年的工作总结到一起然后发一篇,做里程碑式的工作,也给其他学者留了优化和改进的空间。



















 1948
1948

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











