







背景
表观基因组的调控异常驱动了转录程序的异常,从而促进了癌症的发生和发展。基因调控缺陷除了经常影响致癌和肿瘤抑制网络之外,参与抗肿瘤反应的肿瘤免疫原性和免疫细胞也可能受到表观基因组改变的影响。这一发现对表观遗传疗法和癌症免疫疗法及其组合疗法的开发和应用具有重要意义。这篇综述回顾了DNA甲基化和组蛋白翻译后修饰这两种关键的异常表观遗传过程在肿瘤免疫原性中的作用,以及表观遗传调节对抗肿瘤免疫细胞功能的影响。还强调了调节表观遗传的小分子抑制因子在增强抗癌免疫反应中的作用,并讨论了利用癌症表观遗传学和癌症免疫学之间复杂的作用关系来开发表观遗传疗法和免疫疗法相结合的治疗方案所面临的挑战。
简介
2020年12月14日,来自Department of Oncology, The University of Melbourne, Parkville, Victoria, Australia等机构的Simon J. Hogg、Paul A. Beavis、Mark A. Dawson和Ricky W. Johnstone 在Nature reviews. Drug discovery(IF: 64.797)杂志上发表题为“Targeting the epigenetic regulation of antitumour immunity”的综述[1]。文章回顾了针对表观遗传过程的抗癌药物的两个关键领域:影响肿瘤免疫原性的肿瘤细胞对药物的反应;以及直接参与抗肿瘤反应的免疫细胞对药物反应。还讨论了如何理解癌症表观遗传学和癌症免疫学之间的相互作用,为开发表观遗传学药物和免疫疗法相结合的新治疗方案提供基础。
主要内容
表观遗传药物对肿瘤细胞的作用
DNA甲基化调节因子在肿瘤发生和肿瘤免疫原性中的作用。小分子抑制因子DNA甲基转移酶(DNMT),俗称去甲基化药物,是治疗癌症最广泛使用的表观遗传学疗法,主要用于治疗骨髓增生异常综合征(MDS)和急性髓系白血病(AML)。5-Azacitidine (5-Aza), 5-aza-2′-deoxycytidine (decitabine)和SGI-110 (guadecitabine)是胞嘧啶核苷的类似物,它们不可逆地将DNMT蛋白结合到DNA上,导致DNA低甲基化。DNMT抑制剂诱导的直接抗肿瘤作用,如细胞凋亡、细胞周期阻滞和分化,被归因于DNA甲基化沉默的基因的重新表达。原发肿瘤的基因表达谱表明,在使用DNMT抑制剂和去甲基化药物治疗后,免疫相关途径的增加可以提高与抗原提呈相关的基因的表达,以及CD80、CD86和CD40等免疫刺激因子的表达。
重要的是,并不是所有由去甲基化药物引起的免疫学变化都被认为促进了抗肿瘤反应,因为免疫检查点(PDL1、PD1、PDL2和CTLA4)的有效上调与MDS、慢性粒单核细胞白血病或AML患者对5-Aza的治疗耐药性有关。但是,将DNMT抑制和免疫治疗结合起来,以诱导更有效的抗肿瘤免疫反应,并颠覆与免疫检查点相关的获得性免疫抵抗,或许是一个可行的策略。为此,将DNMT抑制剂与癌症检查点抑制剂(如抗PD1、抗CTLA4或抗PDL1)相结合的临床试验目前正在大量进行(表1)。
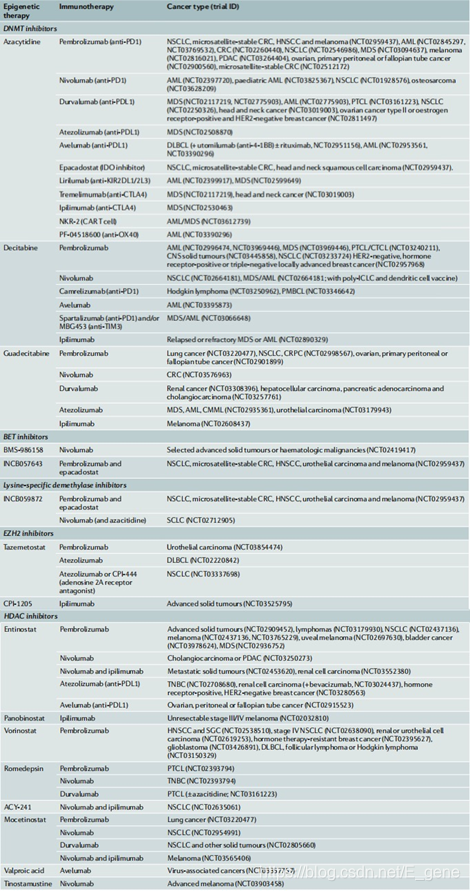
表1.表观遗传疗法与免疫疗法相结合的临床试验
组蛋白乙酰化调节因子在肿瘤发生和肿瘤免疫原性中的作用。蛋白质的翻译后乙酰化是高度动态的,并受组蛋白乙酰基转移酶(HATS)和HDAC的调节。HDAC抑制剂在染色质层次诱导组蛋白和表观遗传调节因子的乙酰化,从而调节RNA聚合酶II(POL II)驱动的转录。在癌细胞中,这种转录调控包括肿瘤发生过程中沉默的基因的重新表达。
在过去的20年里,HDAC抑制剂作为抗癌药物得到了广泛的研究,30项针对实体瘤中HDAC抑制剂的独立临床试验的回顾性分析显示,对HDAC抑制剂的治疗反应取决于联合方案的选择。目前限制HDAC抑制剂在实体瘤中疗效的机制尚不清楚,但总的来说,HDAC抑制同时调节肿瘤细胞免疫原性的正负调节因子、先天免疫系统和获得性免疫系统的识别。HDAC抑制剂的免疫调节活性为其配合免疫治疗提供了一定的理论基础。而HATS虽然在临床开发上不如HDAC抑制剂先进,但其用于癌症治疗的治疗靶点目前正在被评估。
组蛋白甲基化调节因子在肿瘤发生和肿瘤免疫原性中的作用。组蛋白甲基化可能会对转录输出产生相反的影响,这取决于被修饰的特定组蛋白尾部残基和甲基化化学计量比(例如,单甲基化、双甲基化或三甲基化)。这些“癌组蛋白”突变主要抑制组蛋白甲基转移酶(HMT)的活性,促进恶性转化。一些HMT已被当作治疗靶点,包括zeste homologue 2增强子(EZH2), SET domain bifurcated 1 (SETDB1)和disruptor of telomeric silencing 1-like (DOT1L);然而,只有EZH2和DOT1L的小分子抑制剂进入了临床开发。EZH2在机制上与多种肿瘤类型的免疫逃避有关,最显著的是通过抑制MHC I类抗原提呈。
到目前为止,很少有组蛋白去甲基化酶被用作抗癌靶点。赖氨酸特异性去甲基化酶1(LSD1/KDM1A)是H3K4和H3K9的FAD依赖性去甲基化酶,是唯一一种小分子抑制剂进入临床应用的组蛋白去甲基化酶。LSD1能有效沉默基因组中ERV元件的转录,可能通过多种不同的机制调节肿瘤免疫反应,这些机制可能被用来推动抗肿瘤治疗。
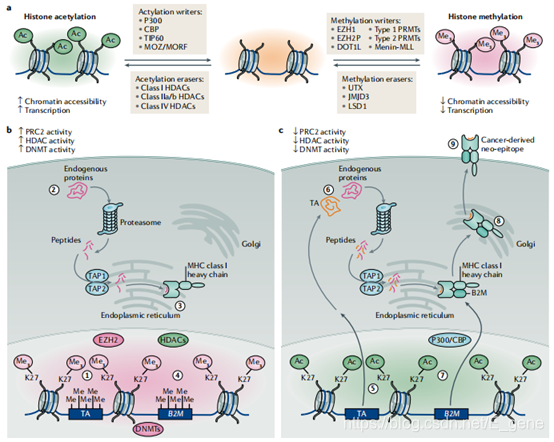
图1. 组蛋白乙酰化和甲基化在肿瘤发生和免疫原性中的作用
赖氨酸乙酰化BET readers在肿瘤发生和肿瘤免疫原性中的作用。溴域是组蛋白和非组蛋白上乙酰化赖氨酸残基的表观遗传识别工具。溴结构域和末端外(BET)蛋白在活性增强子和启动子区域处富集,并在功能上与转录辅助激活因子结合,积极调节RNA Pol II依赖的转录。针对溴域的BET抑制剂已经针对一系列疾病进行了研究,其中几种已经进入临床试验。BET抑制剂在遗传多样性的实体和血液肿瘤中也显示出广泛的疗效。BET抑制剂的抗肿瘤作用源于抑制Pol II驱动的致癌转录,尤其是在血液系统恶性肿瘤中最显著的抑制MYC和MYC依赖的转录程序。尽管BET蛋白在基因组中无处不在地占据活跃的顺式调节元件,但这种富集本身并不能预测对BET抑制剂转录抑制的易感性。BET抑制剂的基因特异性效应可能源于药物的可及性和BET蛋白在不同基因组位置的结合方式。最近的临床前数据表明,BET抑制剂可以增强癌症免疫治疗的活性,BET溴域抑制剂与宿主免疫系统接触,这可能被用来促进抗肿瘤免疫反应。这些研究强调,除了靶向沉默基因表达的机制外,促进选择性转录抑制的表观遗传疗法也可以增强抗肿瘤免疫力。
表观遗传药物对免疫细胞的影响
虽然表观遗传疗法发展的主要原理是利用肿瘤细胞的内在依赖性,但它们在癌症背景下对免疫系统的功能也必须产生一定的影响。这一部分强调了表观遗传机制如何在抗肿瘤免疫反应背景下控制免疫细胞亚群的活力和功能,重点放在了CD4+和CD8+T淋巴细胞和NK细胞上。
NK细胞的表观遗传调控。自然杀伤细胞(NK细胞)是天然免疫的重要媒介,具有很强的细胞毒性,是抗肿癌免疫必不可少的细胞因子。NK细胞功能的调节是通过一系列受体-配体相互作用介导的,最终由正负激活信号(如NKG2DL和MHC-I类)的相对比例控制。激活后,NK细胞成熟,逐渐获得抗肿瘤功能,包括直接的细胞毒活性和细胞因子的产生。
研究表明,表观遗传疗法可以用来调节NK细胞的功能,从而增强抗肿瘤免疫。这一效应具有潜在的重要性,因为除了直接的抗肿瘤细胞毒作用外,NK细胞还需要通过募集常规的1型树突状细胞来获得最佳的抗肿瘤T细胞反应。表观遗传疗法对NK细胞介导的抗肿瘤免疫的直接作用(expression of IFNγ和NKG2D by NK cells)和间接作用(expression of NKG2DL, MHC class I 和 CXCL9/CXCL10)之间具有复杂的相互作用关系,在同基因模型中进一步研究表观遗传对NK细胞介导的抗肿瘤反应的作用是有先例的。
CD4+T细胞的表观遗传调控。CD4+辅助性T细胞分为不同的亚群,包括TH1细胞、TH2细胞、产生IL-17的辅助性T细胞(TH17)、胸腺来源的Treg细胞(tTreg细胞,形成于胸腺)、外周来源的Treg细胞(pTreg细胞,形成于外周)、T滤泡辅助细胞(TFH)、TH22细胞和TH9细胞,其特征是具有谱系特异性的转录因子。在肿瘤中,TH1细胞的存在通常与积极的抗肿瘤免疫反应和改善患者预后有关,这是因为TH1细胞具有产生促炎细胞因子和促进CD8+抗肿瘤T细胞反应的能力。相反,FOXP3+Treg细胞和GATA3+TH2细胞在功能上抑制TH1细胞的反应,从而促进肿瘤生长。辅助性T细胞系的分化是可塑性的,在适当的环境刺激下可以逆转,这一过程与表观遗传和转录的动态变化有关。因此,与活性转录相关的组蛋白修饰,如H3K4me3,在Treg细胞上的效应器相关基因,如IFNG和CD154(编码CD40L),与传统的Teff细胞相比没有那么丰富。
总体而言,谱系特异性的CD4+良好的表观遗传依赖性可能有助于形成最佳的CD4+T细胞反应。鉴于Treg细胞和CD4+Teff细胞在抗肿瘤免疫中的重要性,有必要进一步研究通过靶向特定的表观遗传复合物来操纵CD4+T细胞向有利的抗肿瘤表型转化的可能性。
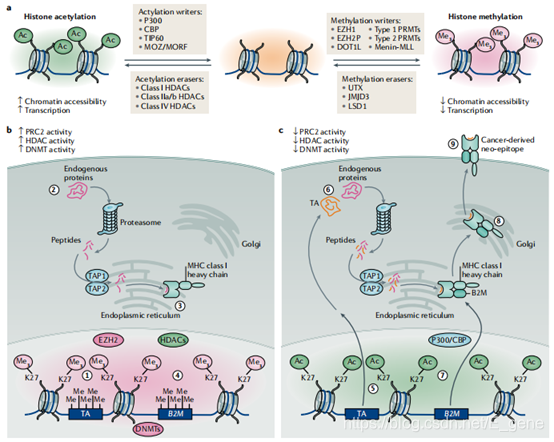
图2. NK细胞、CD4+T细胞和Treg细胞抗肿瘤活性的表观遗传调控。
CD8+T细胞的表观遗传调控。由于CD8+T细胞在抗肿瘤免疫反应和病毒清除中的重要作用,CD8+T细胞的表观遗传调控已被广泛研究。在T细胞分化的线性模型下,T细胞逐渐分化成被描述为干细胞记忆T(TSCM)细胞的表型,然后是中央记忆T(TCM)细胞、效应记忆T细胞、Teff细胞,最后是耗尽的T细胞或终末分化细胞。对CD8+T细胞发育和耗竭的时间评估表明,染色质的可及性、增强子的使用和转录因子的结合是很明显的,特别是在涉及T细胞分化的关键基因,由此可以预见,CD8+T细胞激活会导致DNA和组蛋白修饰的动态变化。
DNA甲基化积极调节T细胞分化和效应器功能,并限制对免疫检查点阻断的治疗反应,DNA甲基化增强的转录抑制在PD1等免疫检查点被阻断后是不可逆的,这表明它限制了CD8+T细胞对PD1阻断的反应性。因此,将抗PDL1、抗PD1或抗CTLA4与DNMT抑制剂联合使用可产生更强的CD8+T细胞抗肿瘤免疫应答。目前,结合甲基转移酶抑制剂和免疫检查点抑制剂的临床试验现已开始。
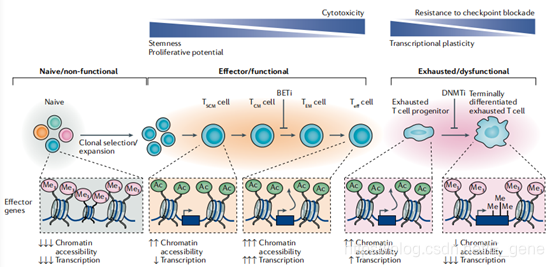
图3.CD8+T细胞分化和耗竭过程中效应器功能的表观遗传调控
对免疫治疗的启示
调节内源性抗肿瘤免疫。当考虑癌症免疫治疗时,了解TME内CD8+T细胞的表观遗传调控是至关重要的。如上所述,CD8+肿瘤浸润性淋巴细胞的特征是具有增强子、染色质结构和甲基化模式的,这限制了抗肿瘤免疫反应的持久性。因此,终末分化耗竭T细胞的效应功能不能被检查点阻断(如抗PD1或抗CTLA4)完全挽救,因为不可逆的表观遗传状态会带来的基因抑制。初始T细胞的TCR活化通过活化T细胞核因子(NFAT)与辅因子(AP1)的功能相互作用而导致基因表达。然而,在衰竭的T细胞中,TCR的激活(或对PD1阻断的反应)会导致NFAT反应与无效反应相关的基因选择性增强。这就解释了为什么与非终末分化的CD8+T细胞不同,记忆前体TCF7+细胞,也被称为前体耗竭T细胞,含有激活的组蛋白修饰和DNA低甲基化,并且对检查点阻断反应最快。因此,未来的努力应该集中于确定操纵CD8+T细胞的策略,以形成更容易适应免疫检查点阻断的强大和持久反应的表观遗传学特征。
过继细胞治疗的调节。表观遗传疗法的一个令人兴奋的治疗应用在于它们与过继细胞疗法(ACT)的结合使用,包括嵌合抗原受体T细胞(CAR T细胞)疗法。ACT的制造和输送过程允许肿瘤在ACT之前接受表观遗传疗法,或T细胞在回输之前接受表观遗传疗法的预处理。表观遗传疗法(如EZH2、LSD1或HDAC抑制剂)、去甲基化药物以及增加MHC-I类抗原在肿瘤细胞上的表达,为患者提供了预处理的机会,并在T细胞转移之前创造了一个炎症环境。与这一效应一致的是,HDAC抑制剂已被证明可以促进ACT的疗效,这与转移的T细胞向肿瘤部位的运输增加有关。
展望
表观遗传疗法和免疫疗法的结合正在迅速成为癌症治疗的新典范。展望未来,当前表观遗传疗法的特异性和亲和力的提高,以及针对更广泛的表观遗传和免疫学靶点小分子的开发,加上新的下一代测序和免疫学技术,可能会为合理组合治疗方案提供进一步的机会。在单细胞水平上对肿瘤浸润性免疫细胞的转录组进行分析,极大地加深了我们对免疫检查点抑制剂作用机制的理解。同样,我们预计,在单细胞分辨率下对TME中免疫细胞表观基因组的多重分析将导致支持抗肿瘤免疫的基本表观遗传过程的进一步细化。总体而言,利用这些复杂的生物相互作用将为癌症治疗的新发现与改进提供令人兴奋的机会。
参考文献:
- Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020 Nov;19(11):776-800. pii: 10.1038/s41573-020-0077-5. doi: 10.1038/s41573-020-0077-5. PubMed PMID: 32929243.
想要了解更多生信科普小知识,欢迎持续关注我们!


















 6688
6688

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









