







介绍
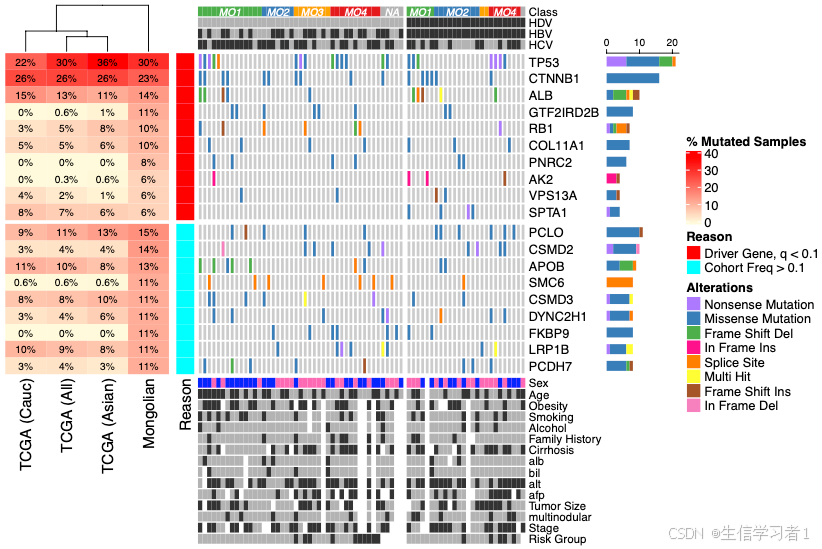
基因突变瀑布图是一种用于展示基因组突变数据的可视化图表,它能够直观地展示每个样本中的突变情况,包括突变类型和突变基因。以下是瀑布图的主要特点和它能说明的问题:
- 展示突变信息:瀑布图的中间主体部分横坐标代表样本,纵坐标代表基因,颜色条块表示该基因在该样本中发生了突变,不同的颜色代表不同的突变类型。
- 突变数目统计:瀑布图最上面的柱状图表示每个样本的突变数目,可以快速了解样本中的突变频率。
- 基因突变类型统计:瀑布图最右侧的条形图表示每个基因不同突变类型的数目,有助于识别特定基因的突变类型分布。
- 样本和基因的排序:瀑布图通过排序可以展示突变个数最多的基因和包含这些基因突变的样本,有助于识别高频突变基因和样本。
- 突变类型的展示:瀑布图可以展示不同类型的突变,如错义突变、移码突变、无义突变、插入缺失等,有助于理解突变的性质和影响。
- 临床信息的关联:瀑布图还可以对样本进行临床信息的注释,观察临床信息与基因突变的相关性趋势。
- 发现潜在的驱动基因:通过识别样本中高频突变的基因,瀑布图有助于发现可能的肿瘤驱动基因。
- 趋势性和互斥伴发现象:瀑布图可以揭示某些基因突变之间的趋势性互斥伴发现象,这对于理解疾病机理和基因相互作用具有重要意义。
数据下载
MongolianHCC数据集:
- 百度网盘链接: https://pan.baidu.com/s/15BE0QMCXKs6VYthZBTZyeQ
- 提取码: dazy
加载R包
library(maftools)
library(TCGAmutations)
library(circlize)
library(ComplexHeatmap)
library(RColorBrewer)
library(openxlsx)
library(dplyr)
代码
PROJECT_DIR <- "./MongolianHCC/" # replace this line with your local path
source(file.path(PROJECT_DIR, "SCRIPTS", "helper_functions.oncoplot.R"))
sample_info_file <- file.path(PROJECT_DIR, "DATA", "PROCESSED", "patient_sample_metadata_w_clustering_risk.txt")
driver_res_file <- file.path(PROJECT_DIR, "DATA", "ORIGINAL", "mut_full.sig_genes.txt")
out_dir <- file.path(PROJECT_DIR, "RESULTS", "mut_oncoplot")
maf_file <- file.path(PROJECT_DIR, "DATA", "PROCESSED", "maf_after_all_filters.maf")
sample_info.exome.file <- sample_info_file
driver_results_file <- driver_res_file
driver_sig_col <- "q"
driver_sig_thresh <- 0.1
driver_sig_freq <- 0.05
cohort_freq_thresh <- 0.2
# cohort_freq_thresh=NA
gene_list_file <- NA
source(file.path(PROJECT_DIR, "SCRIPTS", "helper_functions.oncoplot.R"))
if (!dir.exists(out_dir)) {
dir.create(out_dir, recursive = T)
}
if (grepl(".xlsx$", sample_info.exome.file)) {
library(openxlsx)
sample_info.exome <- read.xlsx(sample_info.exome.file)
} else {
sample_info.exome <- read.table(sample_info.exome.file, sep = "\t", header = T, stringsAsFactors = F)
sample_info.exome$Tumor_Sample_Barcode <- sample_info.exome$WES_T
}
ignoreGenes <- c("TTN", "MUC16", "OBSCN", "AHNAK2", "SYNE1", "FLG", "MUC5B",
"DNAH17", "PLEC", "DST", "SYNE2", "NEB", "HSPG2", "LAMA5",
"AHNAK", "HMCN1", "USH2A", "DNAH11", "MACF1", "MUC17", "DNAH5",
"GPR98", "FAT1", "PKD1", "MDN1", "RNF213", "RYR1", "DNAH2",
"DNAH3", "DNAH8", "DNAH1", "DNAH9", "ABCA13", "SRRM2", "CUBN",
"SPTBN5", "PKHD1", "LRP2", "FBN3", "CDH23", "DNAH10", "FAT4",
"RYR3", "PKHD1L1", "FAT2", "CSMD1", "PCNT", "COL6A3", "FRAS1",
"FCGBP", "RYR2", "HYDIN", "XIRP2", "LAMA1")
# sort(ignoreGenes)
mafObj <- read.maf(maf_file, clinicalData = sample_info.exome)
maf.filter <- mafObj
frac_mut <- data.frame(
Hugo_Symbol = maf.filter@gene.summary$Hugo_Symbol,
frac_mut = (maf.filter@gene.summary$MutatedSamples / as.numeric(maf.filter@summary$summary[3])),
stringsAsFactors = F
)
frac_mut[is.na(frac_mut)] <- 0
# driver_res_file="data/somatic.sig_genes.txt"
# driver_sig_col="q"
# driver_sig_thresh=0.1
# driver_sig_freq=0.05
if (file.exists(driver_res_file)) {
driver_res <- read.table(driver_res_file, sep = "\t", header = T, quote = "", stringsAsFactors = F)
colnames(driver_res)[colnames(driver_res) == "gene"] <- "Hugo_Symbol"
driver_res$FLAG_gene <- driver_res$Hugo_Symbol %in% ignoreGenes
driver_res_plus <- merge.data.frame(driver_res, frac_mut)
driver_res_plus <- driver_res_plus[order(driver_res_plus[, driver_sig_col], decreasing = F), ]
driver_genes <- driver_res_plus$Hugo_Symbol[driver_res_plus[, driver_sig_col] < driver_sig_thresh & driver_res_plus$frac_mut > driver_sig_freq]
} else {
driver_res_plus <- frac_mut
driver_genes <- c()
}
# driver_res_plus <- driver_res
cohort_freq_thresh <- 0.1
if (!is.na(cohort_freq_thresh)) {
freq_genes <- setdiff(driver_res_plus$Hugo_Symbol[driver_res_plus$frac_mut > cohort_freq_thresh], driver_genes)
} else {
freq_genes <- NULL
}
if (file.exists(as.character(gene_list_file))) {
custom_gene_list <- read.table(gene_list_file, stringsAsFactors = F)[, 1]
custom_gene_list <- setdiff(custom_gene_list, c(driver_genes, freq_genes))
} else {
custom_gene_list <- NULL
}
gene_list <- list(driver_genes, freq_genes, custom_gene_list)
reasons <- c(
paste0("Driver Gene, ", driver_sig_col, " < ", driver_sig_thresh),
paste0("Cohort Freq > ", cohort_freq_thresh),
paste0("Selected Genes")
)
genes_for_oncoplot <- data.frame(Hugo_Symbol = c(), reason = c())
for (i in 1:length(gene_list)) {
if (is.null(gene_list[[i]][1])) {
next
}
genes_for_oncoplot <- rbind(
genes_for_oncoplot,
data.frame(
Hugo_Symbol = gene_list[[i]],
reason = reasons[i]
)
)
}
genes_for_oncoplot <- cbind(genes_for_oncoplot,
frac = driver_res_plus$frac_mut[match(genes_for_oncoplot$Hugo_Symbol, driver_res_plus$Hugo_Symbol)]
)
genes_for_oncoplot <- genes_for_oncoplot[!is.na(genes_for_oncoplot$frac), ]
genes_for_oncoplot <- genes_for_oncoplot[order(genes_for_oncoplot$reason, -genes_for_oncoplot$frac), ]
split_idx <- as.character(genes_for_oncoplot$reason)
split_idx <- factor(split_idx, levels = reasons[reasons %in% split_idx])
split_colors <- rainbow(length(levels(split_idx)))
names(split_colors) <- as.character(genes_for_oncoplot$reason[!duplicated(genes_for_oncoplot$reason)])
split_colors <- list(Reason = split_colors)
# source("scripts/helper_functions.R")
oncomat <- createOncoMatrix(maf.filter, g = genes_for_oncoplot$Hugo_Symbol, add_missing = F)$oncoMatrix
write.table(genes_for_oncoplot, file = paste0(out_dir, "/genes_for_oncoplot.txt"), sep = "\t", quote = F, row.names = F)
include_all <- T
if (include_all) {
### createOncoMatrix drops empty samples, so this adds them back in
all_wes_samples <- as.character(sample_info.exome$Tumor_Sample_Barcode[!is.na(sample_info.exome$Tumor_Sample_Barcode)])
extra_samples <- setdiff(all_wes_samples, colnames(oncomat))
empty_data <- matrix(data = "", nrow = nrow(oncomat), ncol = length(extra_samples), dimnames = list(rownames(oncomat), extra_samples))
oncomat <- cbind(oncomat, empty_data)
}
oncomat <- oncomat[match(genes_for_oncoplot$Hugo_Symbol, rownames(oncomat)), ]
onco_genes <- rownames(oncomat)
#### TCGA Comparison Heatmap (from helper_functions.oncoplot.R)
tcga_comparison_results <- make_TCGA_comparison_heatmap(onco_genes, maf.filter, split_at = split_idx)
tcga_comparison_hm <- tcga_comparison_results[[1]]
#### MONGOLIA oncoplot
## Oncoplot parameters
annotate_empty <- ""
annotation_font_size <- 9
annotation_height_frac <- 0.3
onco_width <- 9
onco_height <- NULL
oncomat.plot <- oncomat
colnames(oncomat.plot) <- sample_info.exome$Patient[match(colnames(oncomat.plot), sample_info.exome$Tumor_Sample_Barcode)]
if (is.null(onco_height)) {
onco_height <- max(round(0.2 * nrow(oncomat.plot), 0) + 2, 5)
}
hm_anno_info <- as.data.frame(sample_info.exome[match(colnames(oncomat.plot), sample_info.exome$Patient), ])
rownames(hm_anno_info) <- hm_anno_info$Patient
hm_anno_info <- hm_anno_info[, c(10:ncol(hm_anno_info))]
hm_anno_info$sex <- ifelse(is.na(hm_anno_info$sex), "NA", ifelse(hm_anno_info$sex == 1, "M", "F"))
grouping_var <- "hdv"
group_counts <- c(0, table(hm_anno_info[, grouping_var], useNA = "ifany"))
names(group_counts) <- c(names(group_counts)[2:length(group_counts)], "last")
group_levels <- sort(unique(hm_anno_info[, grouping_var]))
plot_idx <- c()
for (currgroup_idx in 1:length(group_levels)) {
# currgroup_idx=1
currgroup <- group_levels[currgroup_idx]
curr_subset <- hm_anno_info[hm_anno_info[, grouping_var] == currgroup, ]
order_classes <- factor(ifelse(is.na(as.character(curr_subset$class)), "Unknown", as.character(curr_subset$class)))
curr_idx <- orderByGroup(oncomat.plot[, colnames(oncomat.plot) %in% rownames(curr_subset)], order_classes)
plot_idx <- c(
plot_idx,
rownames(curr_subset)[curr_idx]
)
}
plot_order <- plot_idx
################### TOGGLE FOR TOP/BOTTOM ANNOTATION ###################
##### This is for only class/cluster on top
# top_anno_names <- c("Class")
# names(top_anno_names) <- c("class")
# bot_anno_names <- c("Sex","Age","HCV","HBV","HDV","Stage","Tumor Size","Cirrhosis","Obesity","Alcohol","Smoker","Family History","alb","bil","alt","afp","multinodular","Risk Group")
# names(bot_anno_names) <- c("sex","age_bin","hcv","hbv","hdv","stage","tumor_size","cirrhosis","obesity","alcohol","smoker","FHx_LC","alb","bil","alt","afp","multinodular","risk_bin") # column names
################### TOGGLE FOR TOP/BOTTOM ANNOTATION ###################
##### This is for cluster+HDV on top
top_anno_names <- c("Class", "HDV", "HBV", "HCV")
names(top_anno_names) <- c("class", "hdv", "hbv", "hcv")
bot_anno_names <- c(
"Sex", "Age", "Obesity", "Smoking", "Alcohol", "Family History", "Cirrhosis",
"alb", "bil", "alt", "afp", "Tumor Size", "multinodular", "Stage", "Risk Group"
)
names(bot_anno_names) <- c("sex", "age_bin", "obesity", "smoker", "alcohol",
"FHx_LC", "cirrhosis", "alb", "bil", "alt", "afp",
"tumor_size", "multinodular", "stage", "risk_bin") # column names
########################################################################
hm_anno_info <- hm_anno_info[, unique(c(names(top_anno_names), names(bot_anno_names)))]
annocolors <- my_oncoplot_colors(hm_anno_info)
my_types <- unique(unlist(apply(oncomat.plot, 2, unique)))
my_types <- my_types[!my_types %in% c(NA, "", 0)]
col <- c(
Nonsense_Mutation = "#ad7aff",
Missense_Mutation = "#377EB8",
Frame_Shift_Del = "#4DAF4A",
In_Frame_Ins = "#ff008c",
Splice_Site = "#FF7F00",
Multi_Hit = "#FFFF33",
Frame_Shift_Ins = "#A65628",
In_Frame_Del = "#f781bf",
Translation_Start_Site = "#400085",
Nonstop_Mutation = "#b68dfc",
no_variants = "#d6d6d6"
)
top_anno_data <- data.frame(hm_anno_info[, names(top_anno_names)], row.names = rownames(hm_anno_info))
colnames(top_anno_data) <- names(top_anno_names)
bot_anno_data <- data.frame(hm_anno_info[, names(bot_anno_names)], row.names = rownames(hm_anno_info))
colnames(bot_anno_data) <- names(bot_anno_data)
unmutated_annodata <- ifelse(colSums(nchar(oncomat.plot)) == 0, annotate_empty, "")
annocolors$empty <- c("TRUE" = "black", "FALSE" = "white")
top_height <- onco_height * annotation_height_frac * (ncol(top_anno_data) / ncol(bot_anno_data))
top_anno <- HeatmapAnnotation(
empty = anno_text(
unmutated_annodata,
gp = gpar(fontsize = 6, fontface = "bold", col = "grey10"),
location = unit(0.5, "npc"),
which = "column"),
df = top_anno_data,
name = "top_anno",
col = annocolors,
show_annotation_name = T,
na_col = "grey70",
show_legend = F,
simple_anno_size_adjust = T,
height = unit(top_height, "inches"),
annotation_name_gp = gpar(fontsize = annotation_font_size)
)
names(top_anno) <- top_anno_names[names(top_anno)]
bot_anno <- HeatmapAnnotation(
df = bot_anno_data,
name = "bot_anno",
col = annocolors,
show_annotation_name = T,
na_col = "white",
show_legend = F,
simple_anno_size_adjust = T,
height = unit(onco_height * annotation_height_frac, "inches"),
annotation_name_gp = gpar(fontsize = annotation_font_size)
)
names(bot_anno) <- bot_anno_names[names(bot_anno)]
# browser()
names(col) <- gsub("_", " ", names(col))
oncomat.plot <- gsub("_", " ", oncomat.plot)
col_split_idx <- hm_anno_info$hdv
onco_base_default <- oncoPrint(
oncomat.plot,
alter_fun = alter_fun,
col = col,
row_order = 1:nrow(oncomat.plot),
name = "oncoplot",
show_pct = F,
top_annotation = top_anno,
bottom_annotation = bot_anno,
row_split = split_idx,
left_annotation = rowAnnotation(Reason = split_idx, col = split_colors, annotation_width = unit(0.2, "mm")),
row_title = NULL,
column_title = NULL,
column_order = plot_order,
column_gap = unit(0.01, "npc"),
column_split = col_split_idx,
width = 1
)
# browser()
save_name <- paste0(out_dir, "/oncoplot.pdf")
pdf(file = save_name, height = onco_height, width = onco_width)
draw(tcga_comparison_hm + onco_base_default, main_heatmap = 2)
class_labels <- hm_anno_info$class[match(plot_order, rownames(hm_anno_info))]
class_labels[is.na(class_labels)] <- "NA"
class_labels <- factor(class_labels)
slice_labels <- hm_anno_info$hdv[match(plot_order, rownames(hm_anno_info))]
slice_labels[is.na(slice_labels)] <- "NA"
slice_labels <- factor(slice_labels)
myclasses <- split(class_labels, slice_labels)
label_idx <- lapply(myclasses, function(x) {
# x=myclasses[[1]]
change_idx <- c(which(as.logical(diff(as.numeric(x)))), length(x))
change_idx <- change_idx / length(x)
curr_label_idx <- rep(1, length(change_idx))
for (i in 1:length(change_idx)) {
prev_start <- ifelse(length(change_idx[i - 1]) > 0, change_idx[i - 1], 0)
curr_label_idx[i] <- (change_idx[i] - prev_start) / 2 + prev_start
}
mynames <- ifelse(diff(c(0, change_idx)) < 0.1, "", levels(x))
names(curr_label_idx) <- mynames
return(curr_label_idx)
})
## Add cluster labels to top annotation
for (slice_num in 1:length(label_idx)) {
for (class_num in 1:length(label_idx[[slice_num]])) {
labeltxt <- names(label_idx[[slice_num]])[class_num]
labelpos <- label_idx[[slice_num]][class_num]
decorate_annotation("Class", slice = slice_num, {
grid.text(labeltxt, labelpos,
0.5,
default.units = "npc", gp = gpar(fontsize = annotation_font_size * 0.9, fontface = "italic", col = "white")
)
})
}
}
dev.off()
#### TABLE containing data for each variant
output_data <- maf.filter@data[maf.filter@data$Hugo_Symbol %in% onco_genes, ]
output_data$tumor_genotype <- apply(output_data[, c("Tumor_Seq_Allele1", "Tumor_Seq_Allele2")], 1, paste, collapse = "/")
output_data$normal_genotype <- apply(output_data[, c("Match_Norm_Seq_Allele1", "Match_Norm_Seq_Allele2")], 1, paste, collapse = "/")
pheno_info <- sample_info.exome[match(output_data$Tumor_Sample_Barcode, sample_info.exome$Tumor_Sample_Barcode), ]
pheno_info <- cbind(pheno_info[, "Tumor_Sample_Barcode"], pheno_info[, -c("Tumor_Sample_Barcode")])
pheno_columns <- colnames(pheno_info)
names(pheno_columns) <- make.names(pheno_columns, unique = T)
output_data <- cbind(output_data, pheno_info)
cols_for_table <- c(
"Hugo Symbol" = "Hugo_Symbol",
"Variant Classification" = "Variant_Classification",
"Variant Type" = "Variant_Type",
"Consequence" = "Consequence",
"Chromosome" = "Chromosome", "Start Position" = "Start_Position", "End Position" = "End_Position", "Strand" = "Strand",
"Reference Allele" = "Reference_Allele",
"Tumor Genotype" = "tumor_genotype",
"Normal Genotype" = "normal_genotype",
"Transcript Change" = "HGVSc",
"Protein Change" = "HGVSp_Short",
"Normal Depth" = "n_depth",
"Normal Ref Depth" = "n_ref_count",
"Normal Alt Depth" = "n_alt_count",
"Tumor Depth" = "t_depth",
"Tumor Ref Depth" = "t_ref_count",
"Tumor Alt Depth" = "t_alt_count",
"Existing Annotation" = "Existing_variation",
"gnomAD Frequency" = "gnomAD_AF",
"ExAC Frequency" = "ExAC_AF",
"1000Genomes Frequency" = "AF",
"Current Cohort Frequency" = "tumor_freq",
pheno_columns
)
variant_info <- as.data.frame(output_data)[, cols_for_table]
colnames(variant_info) <- names(cols_for_table)
write.xlsx(variant_info, file = paste0(out_dir, "/Table_2.xlsx"))
tcga.white <- tcga_comparison_results[[2]]
tcga.asian <- tcga_comparison_results[[3]]
tcga_lihc_mc3 <- tcga_comparison_results[[4]]
all_dfs <- list(
driver_res_plus,
data.frame(
MONG_frac_mut = (maf.filter@gene.summary$MutatedSamples / as.numeric(maf.filter@summary$summary[3])),
Hugo_Symbol = maf.filter@gene.summary$Hugo_Symbol, stringsAsFactors = F
),
data.frame(
TCGA_All_frac_mut = (tcga_lihc_mc3@gene.summary$MutatedSamples / as.numeric(tcga_lihc_mc3@summary$summary[3])),
Hugo_Symbol = tcga_lihc_mc3@gene.summary$Hugo_Symbol, stringsAsFactors = F
),
data.frame(
TCGA_Asian_frac_mut = (tcga.asian@gene.summary$MutatedSamples / as.numeric(tcga.asian@summary$summary[3])),
Hugo_Symbol = tcga.asian@gene.summary$Hugo_Symbol, stringsAsFactors = F
),
data.frame(
TCGA_White_frac_mut = (tcga.white@gene.summary$MutatedSamples / as.numeric(tcga.white@summary$summary[3])),
Hugo_Symbol = tcga.white@gene.summary$Hugo_Symbol, stringsAsFactors = F
),
data.frame(maf.filter@gene.summary)
)
all_dfs_merged <- all_dfs %>%
Reduce(function(dtf1, dtf2) left_join(dtf1, dtf2, by = "Hugo_Symbol"), .)
write.xlsx(all_dfs_merged, file = paste0(out_dir, "/gene_mutsig_info.xlsx")) # ,asTable = T)
参考
- The genomic landscape of Mongolian hepatocellular carcinoma


















 1020
1020

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











