




 本文详细介绍了基因芯片数据分析的过程,包括质量控制、背景矫正、归一化处理和数据汇总。首先,通过直接观察、简单affy包和affyPLM包进行质量评估;接着,采用RMA和MAS方法去除背景噪声;然后,进行了线性缩放、非线性缩放和分位数归一化;最后,通过统计方法计算基因表达量。整个流程旨在确保数据的准确性和可比性。
本文详细介绍了基因芯片数据分析的过程,包括质量控制、背景矫正、归一化处理和数据汇总。首先,通过直接观察、简单affy包和affyPLM包进行质量评估;接着,采用RMA和MAS方法去除背景噪声;然后,进行了线性缩放、非线性缩放和分位数归一化;最后,通过统计方法计算基因表达量。整个流程旨在确保数据的准确性和可比性。
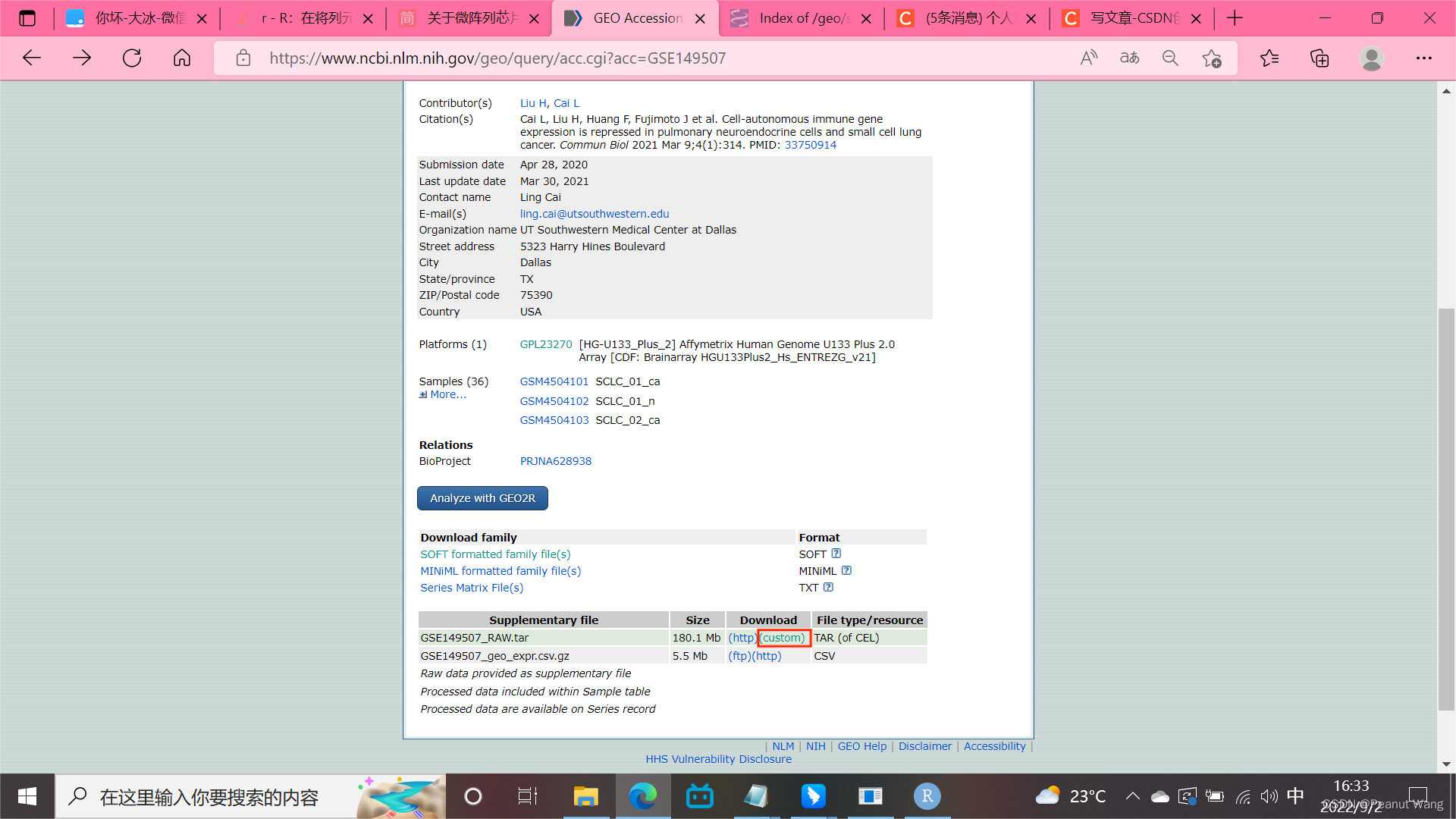
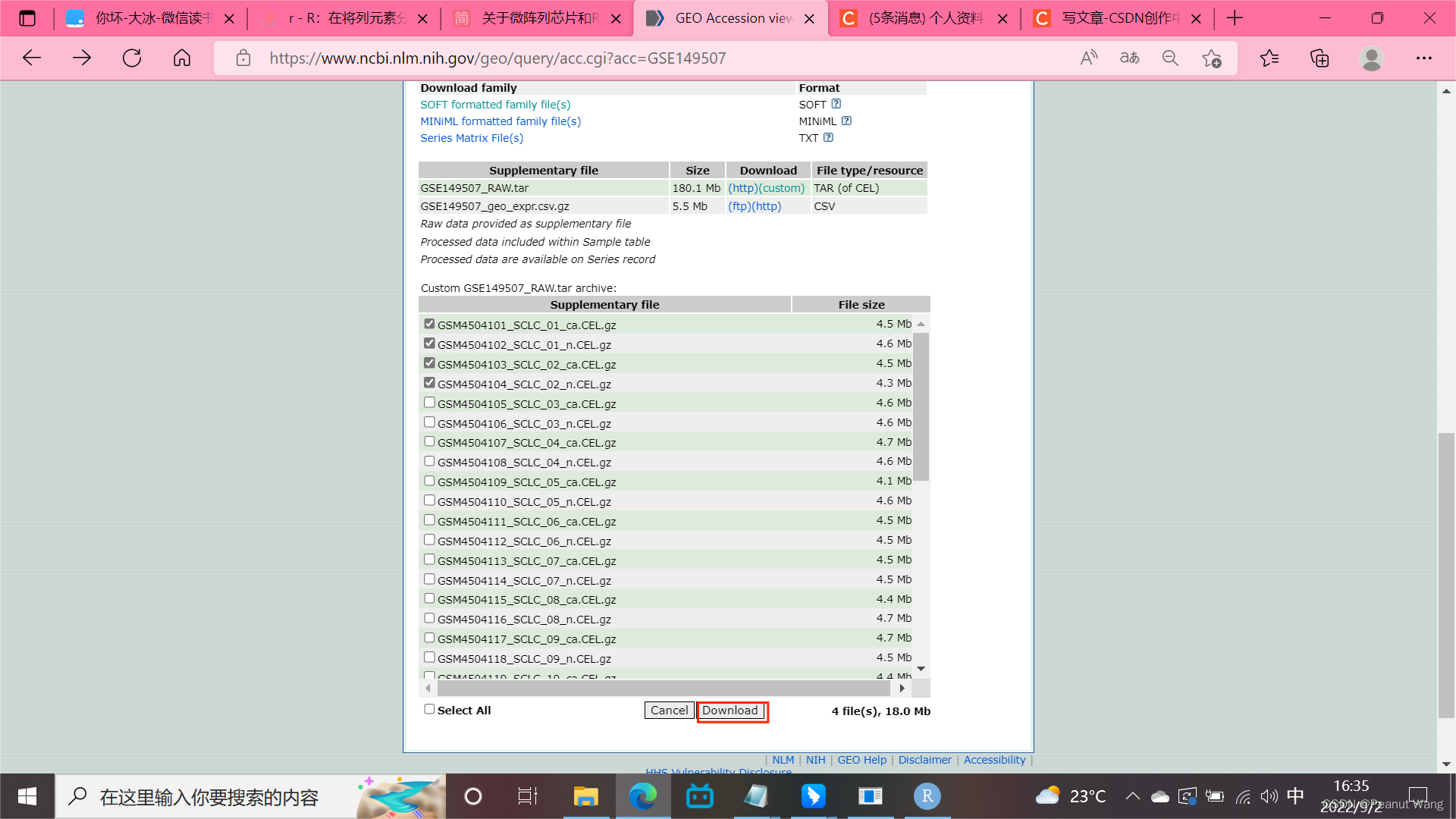
下载四个为例:存放到工作路径中
library(GEOquery)
library(dplyr)
library(tidyverse)
library(affy)
data <- ReadAffy()#读取得到S4对象
第一步:质量控制(直接观察,平均值方法,数据拟合方法分别由image函数,simpleaffy包,affyPLM包实现)
1、直接观察:图像特别黑,说明型号的强度低,图像特别亮,说明信号强度可能过饱和。(在灰度图中不要出现大面积的黑色或白色即可)
# 查看第一张芯片的灰度图像
image(data[,1])
data[,1]
2、simpleaffy包获取质量分析报告,但这个包好像是基于3.0.0以下版本的R软件,所以没有实现。
第一列为所有样本的名称
第二列为检出率和平均背景噪声
第三列蓝色为实现尺度尺子,取值(-3,3),圆形不能超过1.25,否则数据质量不好,三角形不能超过3,否则数据质量不好
bioB说明芯片检测质量没有达标
3、affyPLM包
根据计算结果画权重图,残差图和残差符号图。同样,不要出现大面积异类颜色即可。
library(affyPLM)
Pset <- fitPLM(data)
#### 根据计算结果,
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 4677
4677

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









