






通过以往几期的内容,筛选得到诱饵蛋白的关键互作蛋白之后,通常可以从两方面进行后续的研究:一方面探索蛋白互作的功能,可以通过敲除或过表达互作蛋白后检测下游信号通路或相关表型的变化;另一方面可以探索蛋白互作的机理,即探索这一蛋白互作是通过蛋白的哪些结构域实现的。
目前常见的互作结构域分析流程,一般是通过文献或已有的报道,选择诱饵蛋白和互作蛋白中可能发生互作的结构域,建立截短突变或点突变蛋白,分析蛋白互作的变化。
除了根据已有的相关文献和研究报道判断互作结构域之外,还可以通过以下流程,借助生物信息学工具预测蛋白相互作用的结构域。
1、基于AlphaFold的蛋白结构预测
要预测蛋白的互作结构域,首先需要获得蛋白的结构信息。这类信息可通过PDB数据库查询检索获得,该数据库收录了通过X射线单晶衍射、核磁共振、电子衍射等各种实验手段确定的蛋白质的三维结构。但仍有很多蛋白在PDB数据库中无法找到对应信息或难以准确匹配。这种情况下可通过AlphaFold网站基于蛋白序列预测其结构信息,并下载相应的PDB结构文件。
1) 输入蛋白的Uniprot Accession编号,如Q8IUK8。
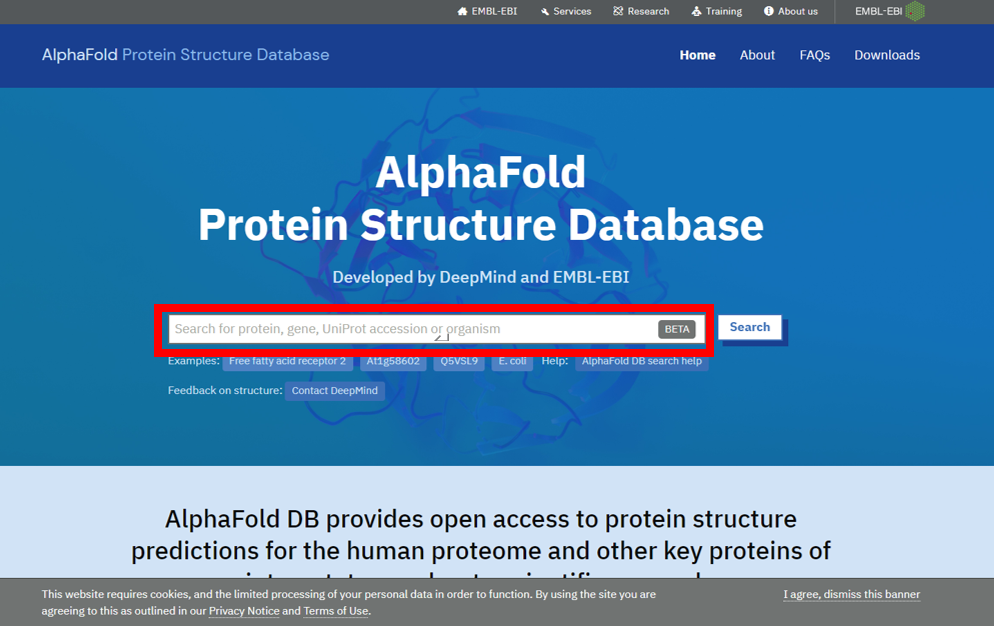
2) 点击进入详情
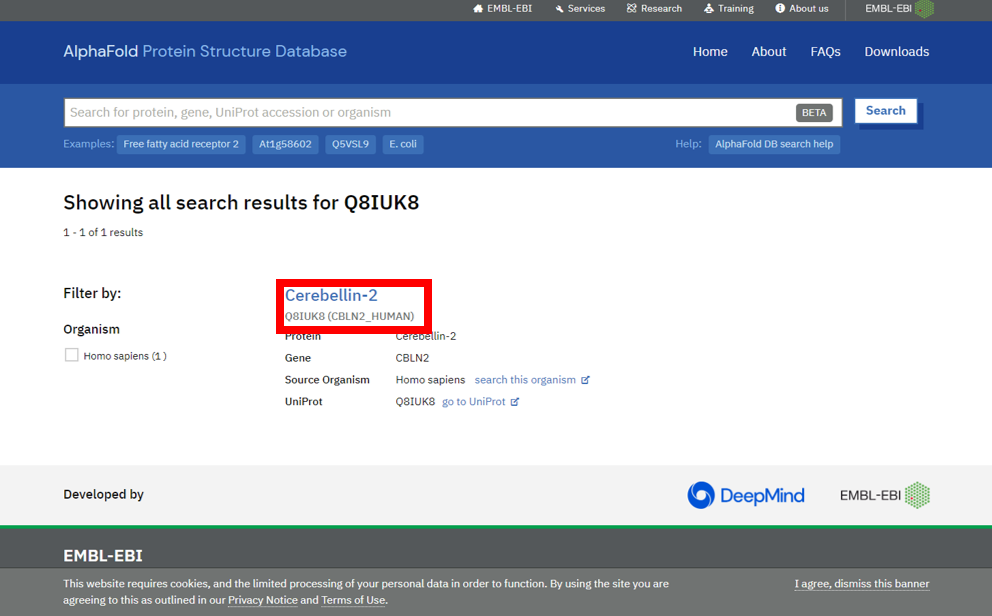
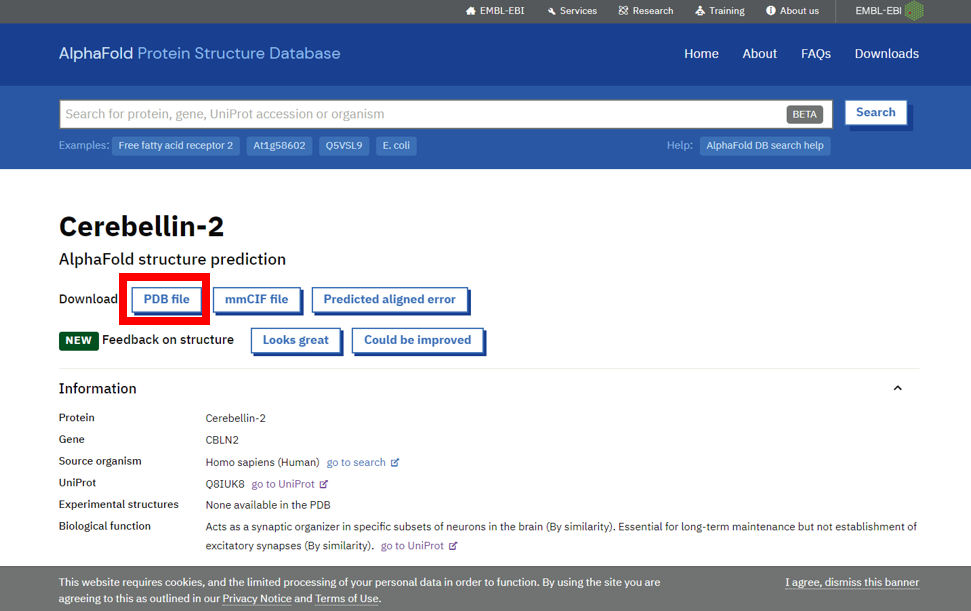
3) 下载PDB结构文件
2、基于Z-dock的蛋白互作分子对接
使用上述方法获得诱饵蛋白和互作蛋白单独的结构数据后,可通过分子对接的方法预测其相互作用形成蛋白复合体的结构。进行蛋白质分子对接预测分析工具有很多,常用的包括RosettaDock、HADDOCK、ClusPro、SwamDock等。这里介绍其中一种Z-dock分子对接的使用方法。
1) 上传PDB结构文件,填写邮箱,提交任务。一般将较大的蛋白作为受体,提交为Protein1。
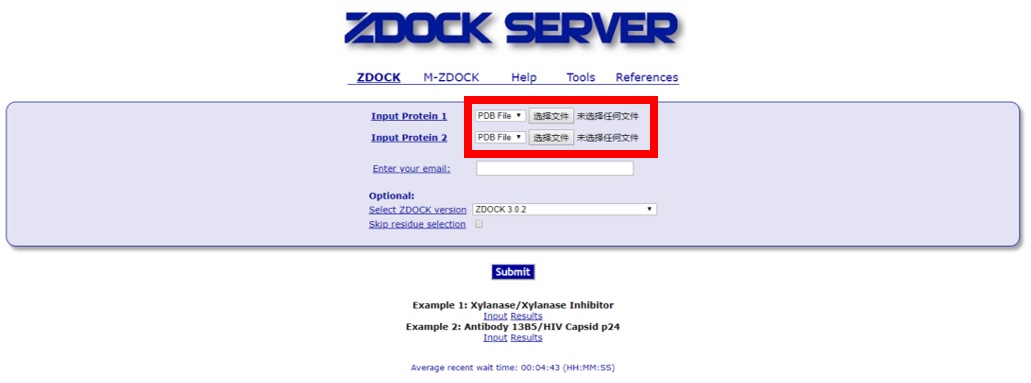
2) 收到的结果链接,下载Top10 predictions,解压获得10个相互作用的PDB结构文件。
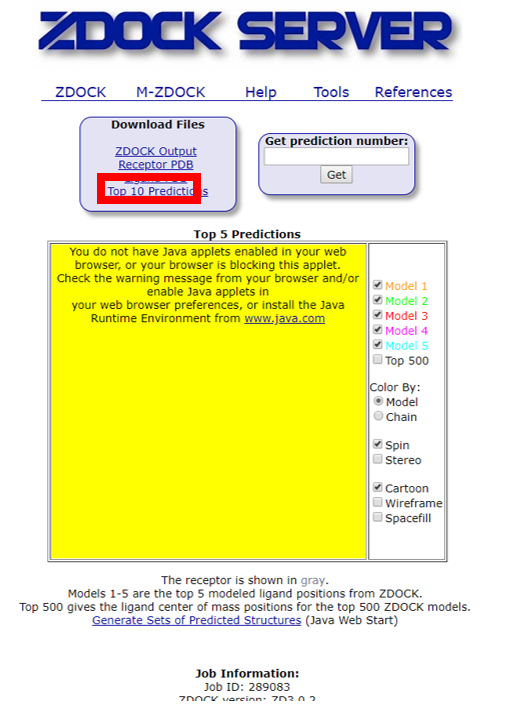
3、基于PDBePISA的蛋白相互作用面分析
通过分子对接获得蛋白复合体结构之后,可以通过Pymol、ChimeraX、Maestro等工具对蛋白复合体三维结构进行可视化。但如果主要目的是分析互作蛋白相互作用的结构域和作用面,使用PDBePISA是一种更简单直接的方法。
1) 上传上一步获得的任一互作PDB结构文件,提交分析任务。
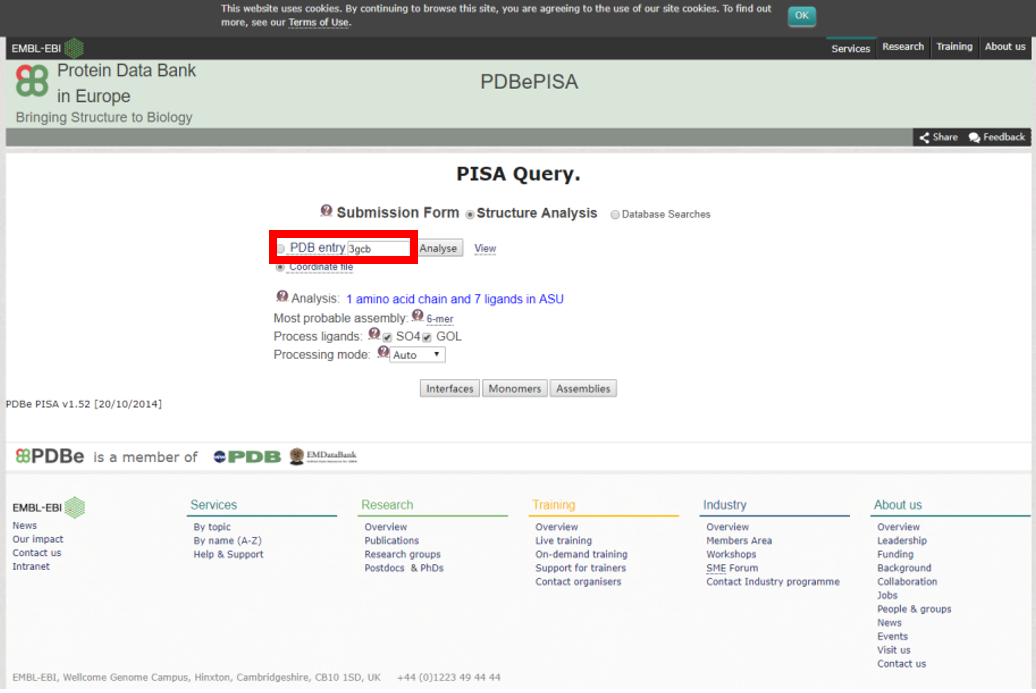
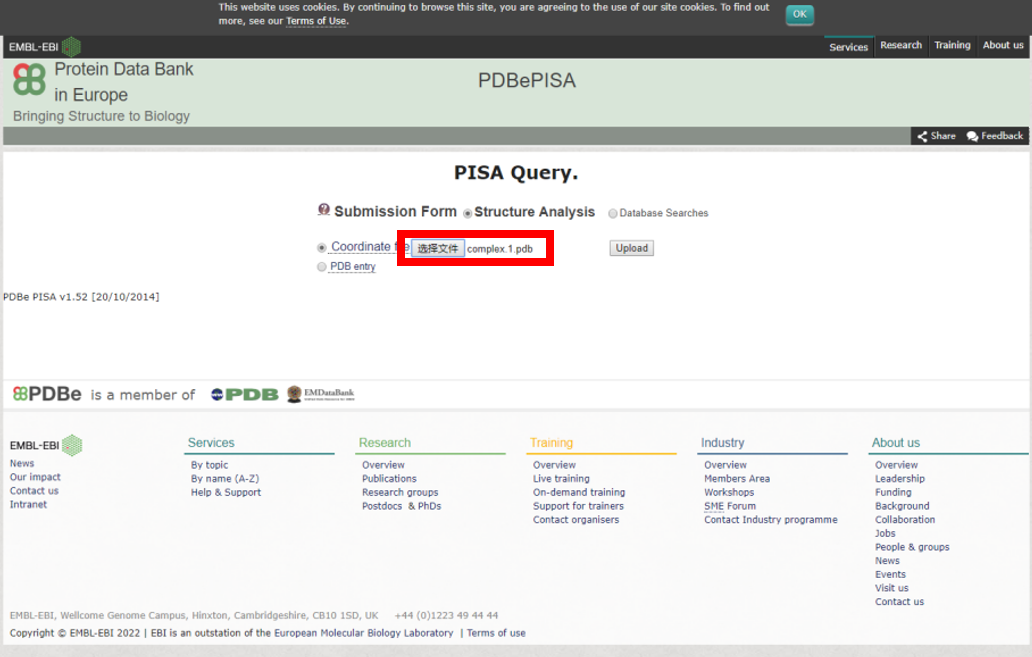
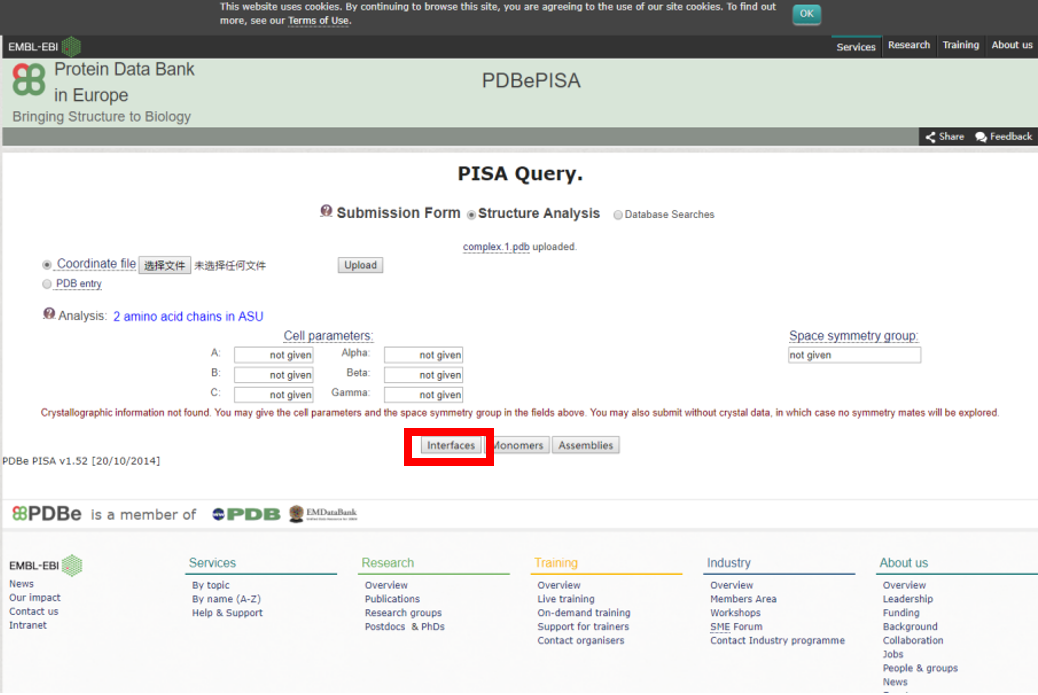
2) 相互作用自由能ΔG<0的互作结构才是有意义的分析结果。查询结果详情。
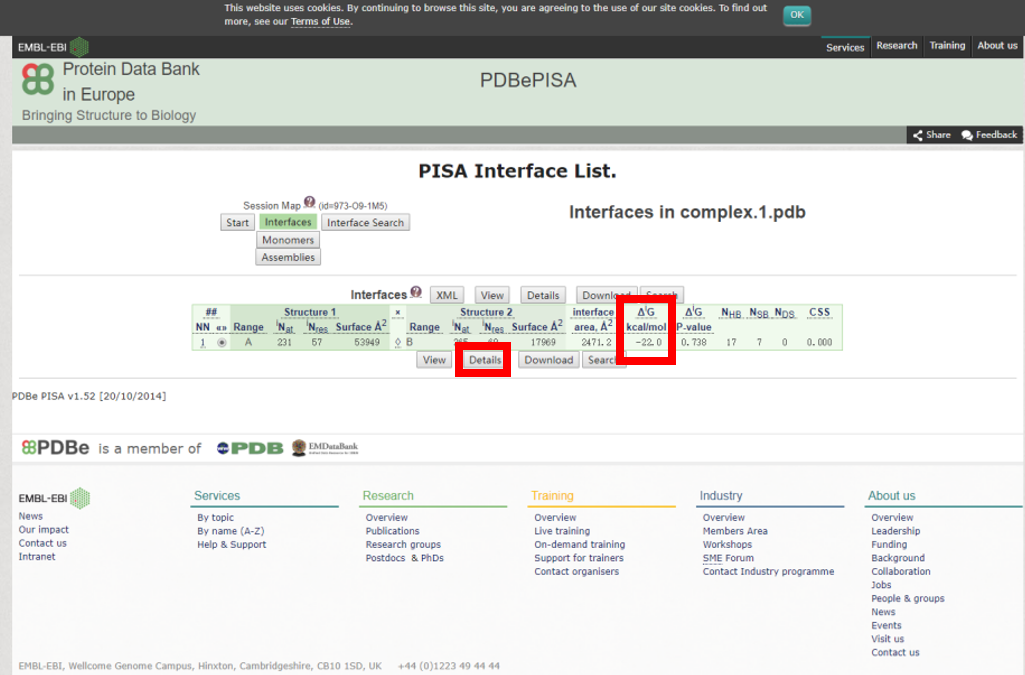
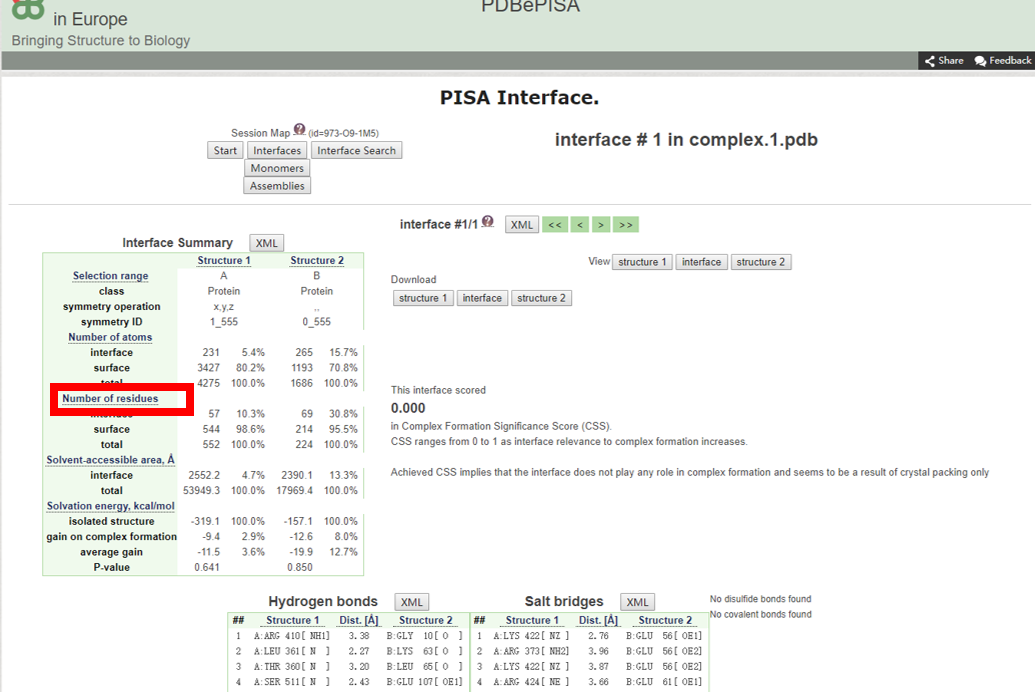
(相互作用的氨基酸残基数)
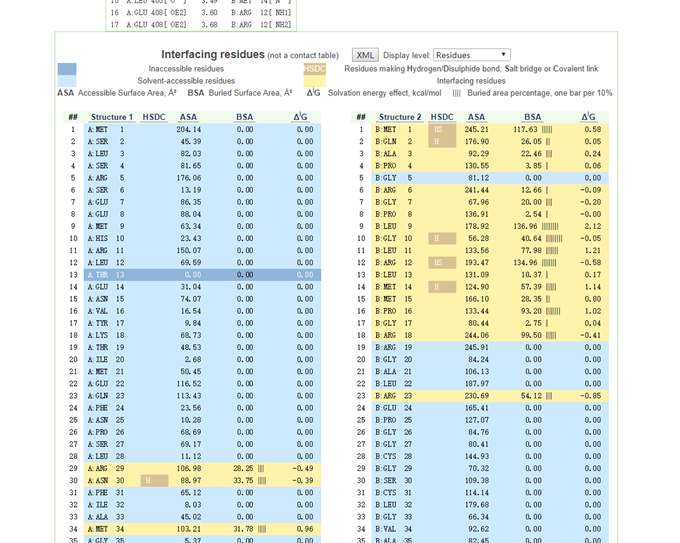
左侧为受体蛋白,右侧为配体蛋白。每一行为一个氨基酸残基,黄色区域为预测结合的区域。HSDC为氢键预测,BSA为互作包埋面积,竖线越多,表明包埋程度越高,可认为互作越紧密。
总结
可通过以下流程分析预测诱饵蛋白和选定互作蛋白相互作用的结构域:
-
通过AlphaFold预测蛋白结构;
-
通过Z-dock预测蛋白复合体结构;
-
通过PDBePISA预测蛋白复合体中蛋白相互作用的区域。
参考文献
-
JUMPER J, EVANS R, PRITZEL A, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature [J], 596: 583-589.
-
PAXMAN J J, HERAS B 2017. Bioinformatics Tools and Resources for Analyzing Protein Structures. Methods Mol Biol [J], 1549: 209-220.
-
PIERCE B G, WIEHE K, HWANG H, et al. 2014. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics [J], 30: 1771-1773.
作者丨尚 骏
审核丨韩强强

















 467
467

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









