







核磁共振波谱法(Nuclear Magnetic Resonance Spectroscopy, NMR )NMR是研究原子核对射频辐射(Radio-frequency Radiation)的吸收。作为现代分析科学的重要技术手段,NMR凭借其卓越的解析能力,成为有机及无机化合物成分鉴定与结构表征的核心工具之一,在定量分析领域也展现其独特价值。该技术已广泛应用于化学、生物、医药等领域的实验室,其应用范围涵盖从简单分子到复杂生物大分子的结构解析。
核磁共振技术在有机分子结构测定中扮演了非常重要的角色,NMR与紫外-可见光谱(UV-Vis)、红外光谱(IR)以及质谱(MS)共同构成现代分析技术的基石,被誉为有机结构解析的"四大名谱"。相较于其他谱学方法,NMR在提供分子空间构型、动态信息及定量氢分布等方面具有独特优势。
然而对于许多学生而言,对NMR的原理、用途以及解析等内容还不是很能理解。本文将系统解析NMR的基本原理、发展历程以及谱图解读方法,开启今天的学习吧!
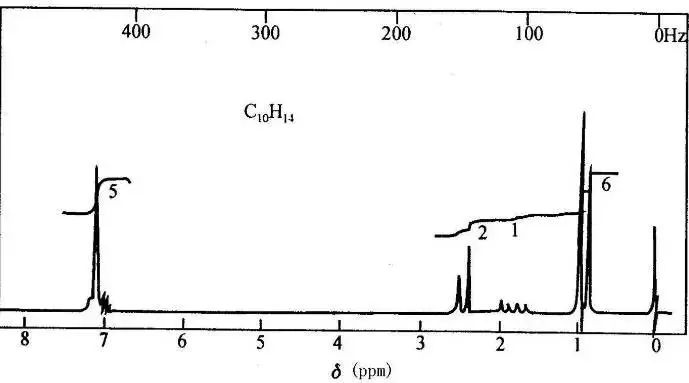
核磁共振谱图
发展历史
咱先来说说NMR的历史发展:
核磁共振(NMR)技术的演进始于20世纪初期的基础物理研究。1924年,奥地利理论物理学家沃尔夫冈·泡利首次提出原子核自旋理论模型,指出特定原子核具备自旋量子数与磁矩特性,其在外磁场中能级分裂的预测为后续研究奠定理论根基。
1946年迎来关键性突破,瑞士物理学家菲利克斯·布洛赫与美国物理学家爱德华·珀塞尔分别通过独立实验,在石蜡和水样本中首次观测到质子核磁共振现象。这项突破性发现使珀塞尔和布洛赫共同斩获1952年诺贝尔物理学奖,此后六十余年间,另有六位学者因推动NMR技术发展荣膺诺奖殊荣。
20世纪50年代标志着NMR技术正式步入分析科学领域,科学家首次成功获取气态氢的核磁共振谱线数据,开启了物质微观结构研究的新纪元。至70年代,该技术实现革命性跨越——磁共振成像(MRI)技术问世,这项创新不仅拓展了NMR的应用边界,更重塑了现代医学诊断范式。
原理
(1)原子核的自旋与磁矩
- 自旋性质:某些原子核(如¹H、¹³C、¹⁹F等)具有自旋角动量,类似于微观的“旋转”。自旋量子数(𝐼I)不为零的核才能产生核磁共振。
- 磁矩产生:自旋的核带有电荷,因此会产生磁矩(𝜇μ),类似一个小磁针。
(2)外加磁场中的能级分裂(塞曼效应)
- 当核被置于强静磁场(𝐵0B0)中时,其磁矩会与磁场相互作用,导致能级分裂。
- 对于自旋量子数 𝐼=1/2I=1/2 的核(如¹H),原本简并的能级会分裂为两个能级:低能级、高能级。
(3)共振吸收(射频脉冲激发)
- 施加一个与能级差(Δ𝐸ΔE)匹配的射频场(射频频率 𝜈ν):
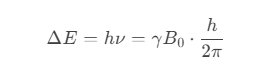
其中,𝛾γ 为核的磁旋比(gyromagnetic ratio),是核的特性常数。
- 当射频频率满足 拉莫尔方程(𝜈=𝛾𝐵0/2𝜋ν=γB0/2π)时,核会吸收能量,从低能级跃迁至高能级,称为 共振。
(4)弛豫与信号检测
- 弛豫过程:射频脉冲停止后,核通过释放能量回到平衡态,分为两种过程:
-
- 纵向弛豫(T₁):恢复磁化矢量的纵向分量(能量释放到周围环境)。
- 横向弛豫(T₂):衰减磁化矢量的横向分量(核自旋间的相互作用)。
- 自由感应衰减(FID):弛豫过程中产生的时域信号被检测线圈接收,形成随时间衰减的FID信号。
(5)傅里叶变换与谱图生成
- 通过傅里叶变换将时域信号(FID)转换为频域谱图,得到不同核的共振频率(化学位移),反映其化学环境差异。
(6)化学位移与结构信息
- 化学位移(δ):由于核周围电子云产生的屏蔽效应,不同化学环境的核实际感受到的磁场(𝐵𝑒𝑓𝑓Beff)不同,导致共振频率偏移。
- 耦合常数(J):相邻核自旋间的相互作用导致谱线分裂,提供分子内相互作用的几何信息。
NMR按被测定对象分为:氢谱1HNMR、碳谱13CNMR、19F、31P及15N等;有机化合物、高分子材料都主要由碳氢组成,所以在材料结构与性能研究中,以1H谱和13C谱应用最为广泛。
NMR按测定样品的状态分为:液体NMR(最常用)、固体NMR(在高分子结构研究中起重要作用)。
用途
除了运用在医学成像检查方面,在分析化学和有机分子的结构研究及材料表征中运用最多。
1. 有机化合物结构鉴定
有机化合物结构解析主要依据核磁共振(NMR)谱学参数进行综合研判。通过化学位移值可辨识特征官能团;依据耦合裂分模式和耦合常数能够推断相邻原子团的连接方式;结合质子信号积分强度则可确定各基团所含质子的比例关系。值得注意的是,NMR技术在化学动力学研究领域具有独特优势。诸如分子内旋转受阻、化学交换反应等动态过程会引起核外电子环境发生特征性变化,这些微观动力学现象会通过谱线线型变化(如峰宽展缩)、信号分裂模式改变或化学位移漂移等特征在NMR谱图中呈现显著响应。
图源:摩熵化学(MolAid)
谱图中化合物的信息
(1)峰的组数:反映分子中磁不等效氢核的种类数目,对应等价质子群的种类数;
(2)峰的强度(面积):表征各类质子的相对数量比例,通过积分曲线高度比实现定量解析;
(3)化学位移值(δ):揭示氢核所处电子云密度的特征参数,与电负性取代基、共轭效应等直接相关;
(4)化学环境标识:通过位移值归属可明确质子所处的特定化学微环境及其空间定位;
(5)裂分模式:根据(n+1)规律判定相邻等价氢的数目(n为邻近等价质子数),解析结构片段连接方式;
(6)偶合常数(J值):反映自旋-自旋耦合作用的强度参数,其数值特征可指认取代基的电子效应、空间排布及分子立体构象。
2. 高分子材料的NMR成像技术
核磁共振(NMR)成像技术在高分子材料领域通过非侵入式三维成像,能够精准检测材料内部缺陷(如微裂纹、空洞),实时监测挤塑成型或发泡过程中分子链的动态行为,优化加工条件;同时可解析粘合剂固化界面作用机制及多孔材料的孔径分布与流体渗透特性,为提升制品性能提供分子层面的结构依据。其结合弛豫时间分析与扩散加权成像,进一步揭示了材料宏观性能与分子运动(如结晶动力学、相分离行为)的定量关联,推动材料设计与工艺创新。
3. 多组分材料分析
核磁共振(NMR)技术在多组分材料分析中通过组分特征信号的独立解析能力,实现复杂体系的精准表征。当聚合物共混物相容性良好时,其分子运动协同性表现为均一的弛豫时间分布,而相容性差则因相分离现象呈现多组分弛豫特征,固体NMR通过弛豫时间分布谱与交叉极化实验可定量评估界面相互作用强度及相区尺寸,为材料结构稳定性与性能优化提供分子动力学依据。尤其值得注意的是,固体魔角旋转(MAS)与多脉冲去耦技术可突破传统表征局限,在解析多相体系时展现高分辨优势。此外,在研究聚合物还用于研究聚合反应机理、高聚物序列结构、未知高分子的定性鉴别、机械及物理性能分析等等。
样品分析
1.样品量不同场强需要的样品量不同
2.氘代试剂的选择
不同的NMR实验需要选择合适的氘代溶剂,主要依据:
- 溶解性:样品必须能充分溶解于溶剂中。
- 化学惰性:避免与样品发生副反应。
- 沸点:影响样品的稳定性和实验温度选择。
- NMR信号特征:某些氘代溶剂仍含有微量¹H,会产生背景峰。
- 价格及可用性:某些氘代溶剂较昂贵,需考虑成本。
3.是否必须加TMS
测试样品加TMS(四甲基硅烷)是作为定化学位移的标尺,也可以不加TMS而用溶剂峰作标尺。
谱图分析与预测
1. 解析核磁共振氢谱
首先确定孤立甲基的类型,通过甲基峰面积的积分高度计算氢原子分布。这一步骤可明确化合物中甲基的数量及所处化学环境。
其次重点解析低场区域的共振吸收峰(如醛基氢δ~9-10、羰基α氢δ~2-3等)。这些特征峰因化学位移范围明确,可通过位移值快速识别对应的官能团类型。
最后处理高级偶合体系。通过分析偶合常数(如J值)、峰分裂模式(如双峰、三重峰)及峰型特征(如AB系统),推测分子中取代基的相对位置、结构异构现象(如顺反异构)以及立体化学信息(如邻位交叉效应)。
2. 解析核磁共振碳谱
首先对比全去偶碳谱线数与分子式碳原子总数。若谱线数等于碳数,说明分子无对称性;若谱线数较少,则表明存在化学环境相同的等价碳,反映分子对称性特征。
随后通过偏共振谱分析各碳信号的分裂情况,确定与其直接相连的氢原子数目(如CH₃呈四重峰、CH₂呈三重峰)。这一步骤可明确碳的级数信息。
最终结合碳化学位移数据库(如羰基碳δ~200-220、芳香碳δ~120-140),归属各碳信号对应的结构单元,尤其关注杂原子邻位碳的显著位移现象。
3. 结合应用碳谱和氢谱
氢谱对无氢官能团(如羰基、氰基)检测存在盲区,而碳谱可精准识别这些基团。例如,在甾体化合物解析中,氢谱常因多个亚甲基化学环境相似产生信号重叠,碳谱却能清晰区分每个碳原子。
碳谱的定量性缺陷(峰高与碳数无正比关系)可通过氢谱的积分定量特性弥补。例如,在确定取代基数目时,可交叉验证氢谱积分面积与碳谱信号数量。
特别对于复杂分子,建议采用"氢谱定位官能团周边环境,碳谱构建整体骨架"的联合解析法,通过二维谱(如HSQC、HMBC)建立碳氢关联网络,实现结构精准解析。
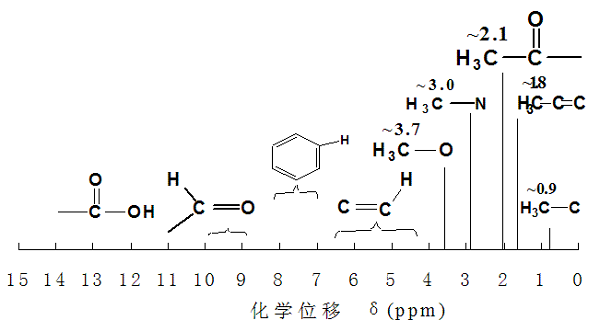
常见类型的有机化合物的化学位移
4. 谱图预测
化学谱图预测在化学分析和生物科学领域中扮演着至关重要的角色。它不仅能够帮助科学家们理解和解释复杂的分子结构,还能在药物设计、疾病标志物的发现以及生物医学研究中发挥重要作用。以核磁共振(NMR)谱图预测为例,其通过模拟化学位移与偶合关系,可精准区分结构异构体(如立体异构、位置异构),显著降低传统实验验证的成本与周期。摩熵化学MolAid平台创新性地实现了谱图反向检索功能,支持用户输入化学位移值反向匹配候选化合物,快速锁定核心结构框架;同时整合权威文献中的实测核磁数据库,通过比对实验值与预测数据的双重验证机制,大幅提升结构确证的可靠性,尤其在天然产物全合成与代谢产物鉴定中展现出高效应用价值。
图源:摩熵化学(MolAid)
5. 如何计算偶合常数?
偶合常数计算需明确其物理定义:自旋偶合导致峰分裂后,相邻两裂分峰之间的频率差(单位Hz)即为偶合常数(J值)。
常见误区纠正:J值≠两组氢的化学位移差值(如—OCH₂CH₃中两组氢δ差>2ppm时,实际计算应取同一组峰内相邻峰的Δδ(ppm)×仪器频率(如400MHz),而非两组峰间差值。例如Δδ=0.5ppm时,J=0.5×400=200Hz)。
结构关联应用:邻位氢(如双键顺式J=6-10Hz,反式J=12-16Hz)通过Karplus公式可计算二面角;远程偶合(如烯丙基J=1-3Hz)反映空间邻近性。

















 1022
1022

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









