







Deep learning models for disease-associated circRNA prediction: a review
 https://academic.oup.com/bib/article/23/6/bbac364/6696465
https://academic.oup.com/bib/article/23/6/bbac364/6696465
![]()
Abstract
新的证据表明,环状 RNA (circRNA)可以为疾病的诊断和治疗提供新的见解和潜在的治疗靶点。然而,传统的生物学实验是昂贵和耗时的。最近,深度学习具有更强大的表示学习能力,使其成为预测疾病相关的 circRNA 的一种有前途的技术。本文主要介绍了目前最流行的与 circRNA 相关的数据库,并总结了基于深度学习的 circRNA 疾病关联预测方法的三种类型: 基于特征生成的方法、基于类型鉴别的方法和基于杂交的方法。对于平衡和不平衡分类任务,我们进一步评价了七个具有代表性的带有真值的平衡和不平衡分类任务的基准模型。此外,我们还讨论了每种方法的优点和局限性,并着重介绍了建议的应用于未来的研究。
关键词: 环状 RNA,circRNA-疾病关联,深度学习,数据库,神经网络
目录
2.1.CircRNA-疾病关联数据库(CircRNA–disease association databases)
2.2.CircRNA注释数据库(CircRNA annotation databases)
3.基于深度学习的计算方法(Deep learning-based computational methods)
3.1.基于特征生成的方法(Feature generation-based methods)
3.2.基于类型分类的方法(Type discrimination-based methods)
3.3.基于混合的方法(Hybrid-based methods)
4.实验评估与分析(Experimental evaluation and analysis)
5.讨论和结论(Discussion and conclusion)
1.引言(Introduction)
环状 RNA (circRNA)是指一种共价封闭形状的新型非编码 RNA,缺乏一个3‘端 ploy-a 尾和一个5’端帽,通过与 RNA聚合酶Ⅱ结合而转录[1]。随着测序技术的进步,人们发现 circRNA 虽然不直接编码蛋白质,但其复杂的功能机制在多种生理过程中起着重要作用。CircRNA 已被发现参与基因表达调控和调节下游信号通路。它也可以像分子“海绵”一样吸收小 RNA (miRNA)。此外,它还作为竞争性内源 RNAs 抑制 miRNA 的功能,并影响相关信使 RNA (mRNA)[2-4]的表达水平等。因此,探索 circRNAs 与 disease 的关联使我们能够在分子分辨率上理解发病机制,并鉴定包括癌症在内的复杂疾病的有希望的生物标志物和治疗靶点。
计算算法的快速发展为研究 circRNA 与疾病的关系提供了新的视角。在这些技术的推动下,研究人员可以在分子水平上研究生物分子的结构和功能,测量患病细胞中的基因表达水平,并鉴定致病基因和易感基因[5,6]。使用计算机算法来预测 circRNA 与疾病的关联,由于其有效性,在疾病诊断和治疗中已经变得越来越突出。事实上,有一些经典的机器学习算法已广泛应用于 circRNA-疾病关联预测任务。例如,Yan 等[7]引入了一个权重递减的 k 最近邻模型来计算关联关系的原始矩阵,并采用基于克罗内克积核的正则化最小二乘方法来推断与疾病相关的 circRNA。Ding 等[8]通过重启随机游走(RWR)提取了相似性网络的特征,并在逻辑回归上推断与疾病相关的 circRNAs。利用四种生物学数据提出了一个基于决策树的梯度提升预测模型。肖等人设计了一个计算模型,通过基于局部不变假设的基于图的多标签学习来发现 circRNAs 与疾病之间的潜在联系。基于重建的 circRNAs 与疾病之间的关联矩阵,魏等[11]应用矩阵分解提取低维特征来解决链接预测问题。Wang 等[12]开发了一种基于推荐算法(PersonalRank)对疾病相关 circRNA 进行分类的计算算法。
在传统的机器学习方法中,有效地获取特征信息对模型性能至关重要。然而,它通常需要专业知识和研究人员的手工设计。多种基于结构的深度学习(DL)算法集成了现有的知识和大规模的数据集,用于数据表示学习。由于其显著的表示学习性能,DL 算法能够以无监督或半监督的方式自动学习具有更多派生信息的特征[13]。此外,一个 DL 模型可以灵活地处理多个任务,并且可以很容易地迁移到不同的知识领域。近年来,DL 已成功地应用于图像识别、自然语言处理等领域[14-16]。DL 的重大进展也促进了 DL 在生物信息学各个领域的应用,例如 DNA/RNA 基序挖掘,单细胞分析和预测 lncRNA (长链非编码RNA)/miRNA 和疾病关联[17-19]。同样,越来越多的基于 DL 的 circRNA 疾病关联预测工具已经证明了它作为一种学习范式的令人印象深刻的能力。
在 circRNA-疾病关联预测的计算模型方面,一些研究者总结了这些模型。例如,肖等[20]和王等[21]主要描述了基于网络算法和基于机器学习的方法。此外,雷等[19]收集了预测疾病与常见非编码 RNA,即 miRNA,lncRNA 和 circRNA 之间关联的计算方法。然而,虽然这些评论介绍了一些 DL 方法,他们没有考虑他们在计算模型的作用的区别。同时,他们没有系统地评价这些方法的性能,以帮助读者根据自己的需要选择正确的方法。因此,本文总结了基于 DL 的预测新型 circRNA 疾病相关性的方法,并将其分为基于特征生成、基于类型鉴别和基于混合的方法三类。And seven representative models were evaluated on benchmark with ground truth for both balance and imbalance classification tasks。
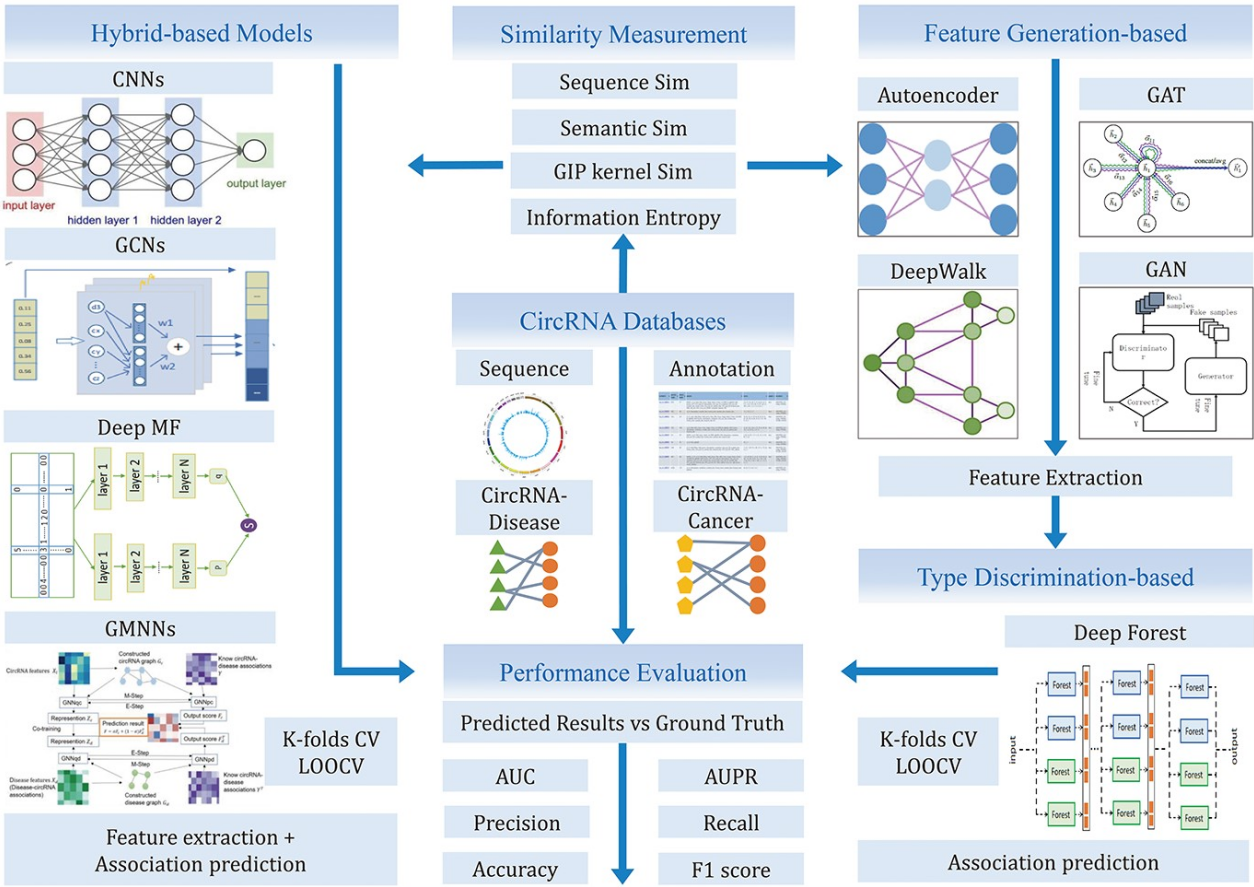
这个回顾的大纲如图1所示。图1分辨率较高的子图载于网上补充数据 http://bib.oxfordjournals.org/。“数据库”部分列出了与本研究相关的各种数据库。“基于深度学习的计算方法”部分介绍了用于 circRNA 疾病关联的三种典型的基于 DL 的预测算法。“实验评价与分析”部分评价和分析了七种具有代表性的方法的性能。在“讨论和结论”部分,对不同类型算法的优缺点以及未来的研究和挑战进行了很好的讨论。
2.数据库(Database)
随着 circRNA 研究的不断深入,生物实验积累了大量的 circRNA 相关数据。一些研究人员收集了它们并建立了相关的数据库[22-34]。这些数据库为基于智能计算方法的 circRNA 疾病关联的预测任务奠定了坚实的基础。在本节中,我们选择几个主要的数据库进行简要介绍。这些数据库可以大致分为两类: circRNA-疾病关联数据库(总结在表1)和 circRNA 注释数据库(总结在表2)。

2.1.CircRNA-疾病关联数据库(CircRNA–disease association databases)
越来越多的证据表明,circRNA 参与疾病的发展和进展,某些 circRNA 被认为是促进癌症进展的功能分子,使其成为有希望的治疗靶点。因此,除了广义的 circRNA- 疾病关联数据库之外,我们还介绍了几个特定的 circRNA- 癌症关联数据库。
典型的 circRNA-疾病关联数据库包括 Circ2Traits [22] ,Circ2Disease [23] ,CircR2Disease [24] ,CircRNADisease [35] ,MNDR 3.0[36]等。如表1所示,Circ2Traits 是人类 circRNA 相关疾病的综合数据库,它通过两种方式研究 circRNA 与疾病的潜在关联。第一种方法是鉴定与疾病和 circRNA 相关的 miRNA。然后计算 miRNA 相关疾病与 circRNA 之间的相关系数。第二种方法是在 circRNA 上注释与疾病相关的单核苷酸突变(SNP) ,然后定位 Ago 蛋白与 circRNA 之间的相互作用位点。后来,研究人员开发了三个数据库,即 CircRNADisease、 Circ2Disease 和 CircR2Disease。在 CircRNADisease数据库中,作者系统地回顾了800多篇与 circRNA 相关的文献,编辑了涉及48种疾病和330种 circRNA 的354个关联条目。在这些疾病中,癌症占54% ,心脑血管疾病占23% 。该数据库还记录了 circRNA 的详细信息,如表达方式、功能描述、宿主基因等注释信息。Circ2Disease 网站提供了5368个经过实验验证的相关关系,包括237个人类 circRNA 和217种疾病。类似于 CircRNADisease 数据库的细节也记录在每个关联中。雷等人2018年发表的 circR2Disease 数据库已被学者广泛用于 circRNA 与疾病的关联预测。他们在2018年3月31日之前检索了 circRNA 相关文献,并编辑了涉及661个人类 circRNA 和100种疾病的739个关联条目。CircR2疾病还包含其他 circRNA 相关记录,如基因符号,circRNA 表达模式(上调或下调) ,实验验证方法(RNAi,QRT-PCR,Northern 和 Western 印迹等)和一些简要描述字段。最近,Ning 等人更新了 MNRD 3.0数据库,该数据库从其他相关数据库整合和发表的文章中收集了296910个实验验证的 circRNA-疾病关系,涉及20506个 circRNA 和1614个疾病。此外,给出了每个关联项的置信度评分。
为了进一步研究癌症特异性 circRNA,学者们建立了几个 circRNA-cancer 关联数据库,包括 CSCD [29] ,CircR2Cancer [37] ,Lnc2Cancer [38]和 MiOncoCirc [2]。CSCD 数据库包含19种癌症类型中的272152个肿瘤特异性 circRNA。在使用 RNA-seq 数据分析 circRNA 期间,使用四种算法,包括 CIRI2,FIND _ CIRC,circRNA _ finder 和 CircExplorer 来鉴定 circRNA。CircR2Cancer 是一个专门用于 circRNA 与癌症关联的数据库,记录了1439个关联记录,包括82个癌症和1135个 circRNA,这些关联记录是通过审查已发表的文献和从 circRNA 疾病关联数据库中筛选而获得的。Lnc2Cancer 数据库包含手工策划和实验支持的人类癌症相关的 circRNA 和 lncRNA。该数据库提供了10303个关联记录,涉及743个 circRNA,2659个 lncRNA 和216种癌症。MiOncoCirc 是第一个根据癌症临床样本编制的数据库,包括2000多个临床肿瘤样本,而其他数据库则基于肿瘤细胞系。MiOncoCirc 的大样本量收集使用户能够进行泛癌分析和直观地了解 circRNA 在各种肿瘤中的表达。
2.2.CircRNA注释数据库(CircRNA annotation databases)
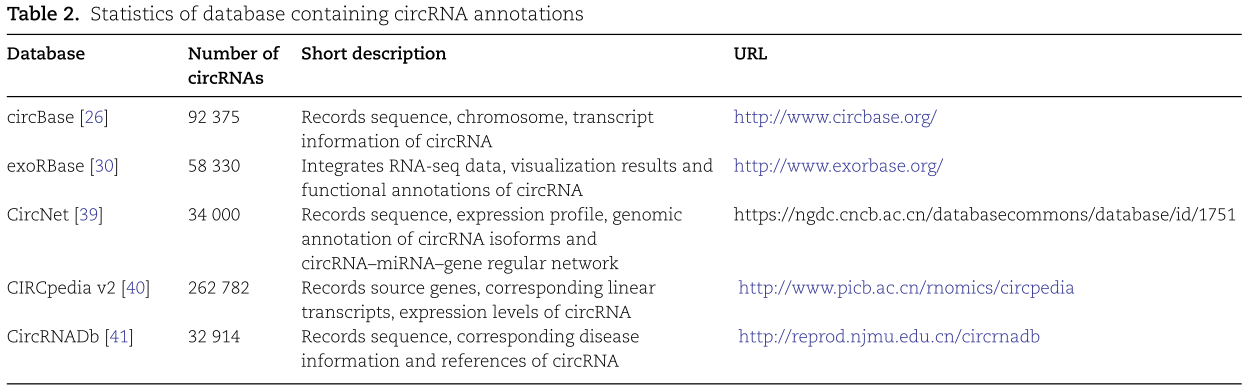
CircRNA 注释数据库提供了大量的注释信息,如序列、表达模式和功能描述等,为学者研究 circRNA 的生物学特性提供了很大的帮助。代表性的数据库是 CircBase [26] ,exoRBase [30] ,CircNet [39] ,CIRCpedia v2[40] ,CircRNADb [41]等。如表2所示,circBase 包含来自人类、小鼠、秀丽隐桿线虫、黑腹果蝇和其他物种的 circRNA,这些 circRNA 来源于已发表的文献和对 circRNA 转录本的注释。通过搜索框可以查询 circRNA 的常见信息,包括序列、染色体、基因组位置、转录本长度和来源样本。该数据库提供各种数据表格的追加下载内容,并允许用户上传自己的演示数据。exoRBase 数据库整合了标准化的人外泌体 RNA-seq 数据和已发表的文献数据,以表征人外泌体中的 circRNA,lncRNA 和 mRNA 表达,以及可视化结果,功能注释和可能的组织起源信息,并鉴定外泌体分子标记。该数据库包含92份血液样本,包含58330个 circRNA,15501个 lncRNA 和18333个 mRNA 数据。CircNet 数据库使用464个 RNA-seq 数据进行新的 circRNA 预测和基因组注释,计算已知和新预测的 circRNA 的表达,最终获得34000个 circRNA。该数据库记录 circRNA 亚型的序列、表达谱和基因组注释,以及 circRNA-miRNA-gene 的规则网络。CIRCpedia 是一个全面的 circRNA 数据库,利用 CIRCExplorer 软件分析了6个物种约180个样本的转录组数据,共鉴定出262782个 circRNA。CircRNA 的 ID,源基因,相应的线性转录本,表达水平,外显子启动和停止位置,细胞系和保守性质可以从这个数据库中检索。CircRNADb 数据库是第一个蛋白质编码的人类 circRNA 数据库,它包含了从相关文献中收集的32914个人类外显子 circRNA。该数据库的特定信息列包括 circRNA 序列、 RNA 剪接序列长度、预测多肽的基本特征、相应的疾病信息和参考文献。
3.基于深度学习的计算方法(Deep learning-based computational methods)
DL 作为传统机器学习方法中的一种有竞争力的方法,在预测 circRNA 与疾病的关联等各种任务中显示出巨大的优势。基于 DL 的计算模型包括以下三个步骤。首先,整合 circRNA 的相似性信息、疾病表型或语义信息以及 circRNA 与疾病的关联信息,构建初始相似性网络。其次,模型基于初始网络提取潜在特征。最后,训练一个分类器来预测疾病和 circRNA 之间的相似性评分。
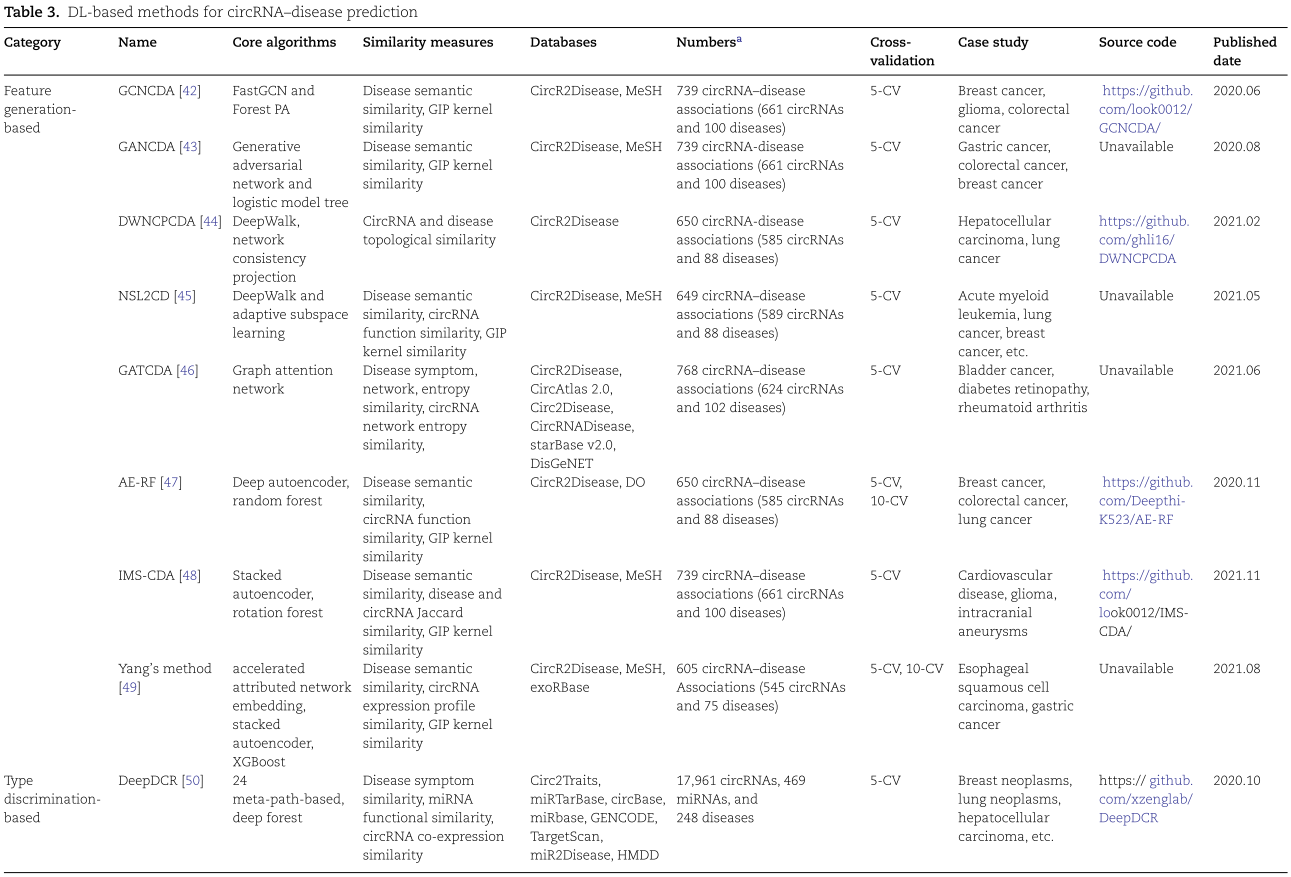
如表3所示,一系列基于 DL 的 circRNA 疾病关联预测方法根据其不同的应用目标分为三类: 基于特征生成的方法、基于类型鉴别的方法和基于混合的方法。对于每个模型,我们主要描述其核心步骤及其优缺点,并详细介绍了它们所使用的 DL 算法。
3.1.基于特征生成的方法(Feature generation-based methods)
具体而言,基于特征生成的方法应用 FastGCN (图卷积网络的快速学习变体)、图注意网络、 DeepWalk、生成对抗网络和基于堆叠自动编码器的算法来描述数据的高阶相关特征。它们通常结合基于多层神经网络的低层特征,形成潜在的高层特征,从而实现无人参与的自动学习特征。然后,应用相应的算法计算与学习特征向量相关联的置信度得分。
3.1.1.GCNCDA
Wang 等[42]通过使用 FastGCN 构建了用于推断潜在 circRNA-疾病关联的 GCNCDA [65] ,应用该方法从数值相似描述器中学习低维和潜在表征。将 circRNA 相似性矩阵 RSim 和疾病相似性矩阵 DSim 水平连接,建立了 circRNA-疾病融合描述器![]() 。相应的公式如下:
。相应的公式如下:
![]()
其中 ![]() 和
和 ![]() 表示 circRNA i 和 disease j。FastGCN 的工作原理是假设当前图是无穷大图的子图,图中的每个节点都是独立同分布的,即
表示 circRNA i 和 disease j。FastGCN 的工作原理是假设当前图是无穷大图的子图,图中的每个节点都是独立同分布的,即![]() 。用卷积方法对连续层进行关联,通过近似积分得到第 l 层的嵌入函数。公式如下:
。用卷积方法对连续层进行关联,通过近似积分得到第 l 层的嵌入函数。公式如下:
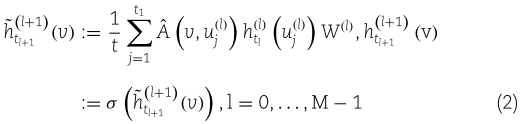
矩阵 ![]() 的元素
的元素 ![]() 的核用
的核用 ![]() 表示,v 和 u 表示两个独立的随机变量。
表示,v 和 u 表示两个独立的随机变量。![]() 分别表示第 l 层的嵌入函数和权矩阵。此外,采用森林-PA (森林惩罚属性)[66]模型来训练 circRNA 与疾病关联的预测分类器。FastGCN 算法不仅通过分层采样加快了 GCN 的训练速度,而且通过重要性采样减小了估计的方差,降低了数据噪声。
分别表示第 l 层的嵌入函数和权矩阵。此外,采用森林-PA (森林惩罚属性)[66]模型来训练 circRNA 与疾病关联的预测分类器。FastGCN 算法不仅通过分层采样加快了 GCN 的训练速度,而且通过重要性采样减小了估计的方差,降低了数据噪声。
3.2.基于类型分类的方法(Type discrimination-based methods)
在基于类型分类的方法中,DL 算法,例如深度森林,被用来训练分类器来执行预测任务的亲和度得分计算。此外,它主要提供了模式分类的判别能力,通常描述了在可见数据条件下后验类别的 circRNA 和疾病关联预测的概率。
3.3.基于混合的方法(Hybrid-based methods)
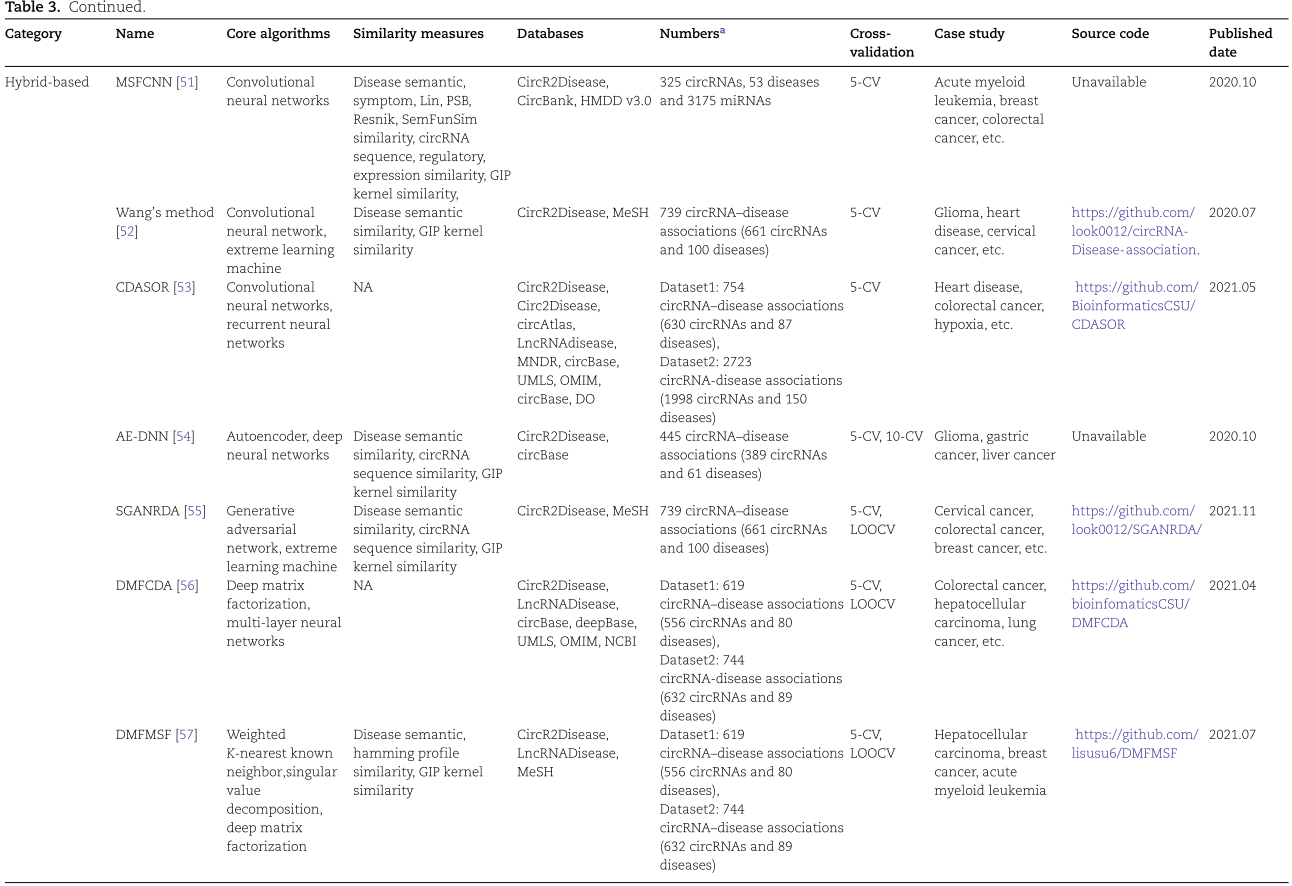
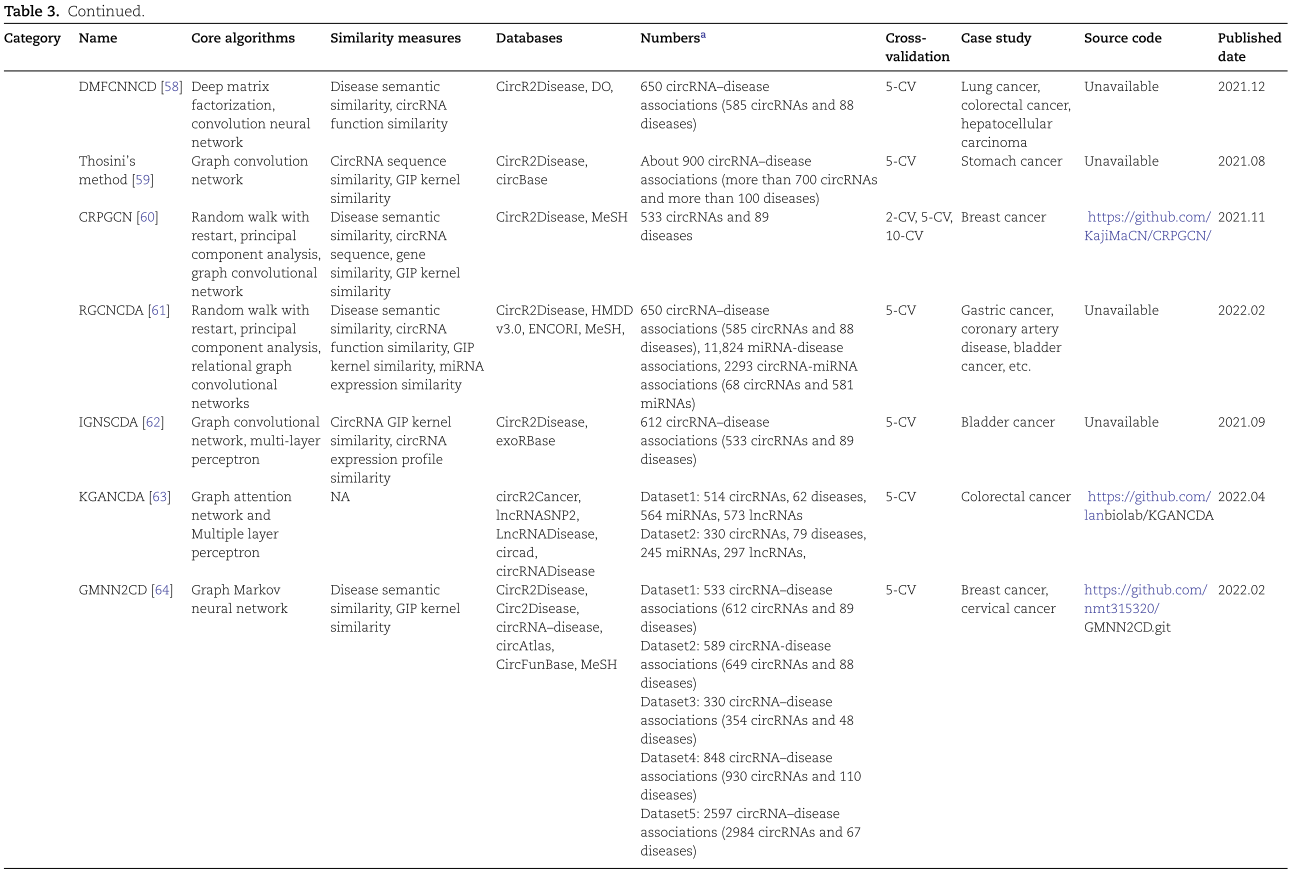
基于混合的方法是将上述特征生成模型与类型识别模型相结合的一种DL模型,如卷积神经网络、深度矩阵分解、图形卷积神经网络和基于图形马尔可夫神经网络的方法。在大多数基于混合方法的模型训练过程中,特征生成模块将模型参数初始化为提取特征的近似最优解,然后反向传播类型识别模块的预测结果与标签之间的损失以调整全局参数,从而解决推断疾病相关 circRNA 的任务。
4.实验评估与分析(Experimental evaluation and analysis)
为了系统地评价关联预测的性能,实验通常按以下步骤进行。在整个过程中,利用重采样的思想,每次选取一部分基准作为模型训练的训练集,并利用剩余的训练集对模型进行验证。通过反复试验,可以得到较为客观的评价结果。典型的策略是留一交叉验证(Leave-One-Out)和 k 折交叉验证。这两种交叉验证策略可以在训练中利用所有可用的样本,同时避免过度拟合。与 k 折交叉验证相比,LOOCV 更适合于小样本的数据集。所得的结果通常用一系列评估指标来量化,包括精确度(precision)、召回率(recall)、 ROC曲线曲线下的面积(AUC)、精确度-召回率曲线下的面积(AUPR)、准确度(ACC)、 f1评分(f1-score)等。最后,病例研究中的几种疾病的调查也是研究人员广泛应用的验证方法。预测数据通常取自用于训练和测试的原始数据集或不同的数据集[101,102]。这些结果是通过对已发表的文献或其他数据库证实的具有高概率分数的 TOP-N 潜在的 circRNA-疾病关联对进行挑选得出的。此外,将生物学实验与算法相结合,验证模型的生物医学真实性,对于发现潜在的疾病相关的 circRNAs 具有重要意义。
在这篇综述中,我们进一步按照上述标准步骤对基于 DL 的 circRNA 疾病关联预测模型进行了全面评估。具体来说,我们进行了七个代表性的涵盖三个类别的 DL 模型,在从 CircR2Disease 数据库中收集的带有真值的金标准基准。去除冗余关联后,我们在585个 circRNA 和88种疾病中获得了613个关联。值得注意的是,有些算法是针对平衡分类问题而设计的,有些则是针对不平衡分类任务而设计的。由于数据类分布的不平衡性,不平衡的分类任务对预测建模提出了挑战。这导致模型的预测性能较差,特别是对于少数群体的分类。为了更公平、更全面地评价和比较这七种方法的性能,我们在基准上分别对平衡和非平衡分类任务执行了七种算法。对于平衡分类任务,从未知关联中随机选取阴性样本,并设置为与阳性样本相同的大小。在七种比较方法中,大多采用了随机阴性抽样策略。为了更完整地再现他们的预测性能,我们遵循了他们的策略,也使用了随机阴性抽样。对于不平衡分类任务,将所有未知关联设置为阴性样本。我们应用5折交叉验证策略对平衡和非平衡数据集上的7个 DL 模型进行评估。七个模型的超参数已在补充表格 s1中总结,该表格已在 http://bib.oxfordjournals.org/上发布。将七个 DL 模型中的每一个预测的 circRNA-疾病关联与真值进行比较。为了量化预测结果,我们绘制了七个 DL 模型的 ROC曲线(ROC)曲线,并计算了它们的 AUC。我们还计算了accuracy分数和 f1分数,并测量了 AUPR。
图7总结了七个基于 DL 的模型的结果。在图7A 中,所有预测模型的 AUC 中值都大于0.7。其中5例 AUC 高于0.9,而 AE-RF 达到最高的 AUC 中值。在图7C 中,DeepDCR 的精度最高,达到0.99,而 RGCNCDA、 AE-RF 和 CPRGCN 的精度值也高于0.9。如图7D 所示,七种方法的 f1得分差异很大,AE-RF 达到0.936,而 CPRGCN 仅为0.15。七个比较算法的预测结果的具体表现,可参阅网上的补充表格 S2( http://bib.oxfordjournals.org/)。总之,AE-RF 对平衡分类任务的预测能力最好,其他四个模型(DMFMSF、 RGCNCDA、 DeepdCR 和 GMnn2CD)的预测能力也相当。
图7总结了七个基于 DL 的模型的结果。在图7A 中,所有预测模型的 AUC 中值都大于0.7。其中5例 AUC 高于0.9,而 AE-RF 达到最高的 AUC 中值。在图7C 中,DeepDCR 的精度最高,达到0.99,而 RGCNCDA、 AE-RF 和 CPRGCN 的精度值也高于0.9。如图7D 所示,七种方法的 f1得分差异很大,AE-RF 达到0.936,而 CPRGCN 仅为0.15。七个比较算法的预测结果的具体表现,可参阅网上的补充表格 S2( http://bib.oxfordjournals.org/)。总之,AE-RF 对平衡分类任务的预测能力最好,其他四个模型(DMFMSF、 RGCNCDA、 DeepdCR 和 GMnn2CD)的预测能力也相当。
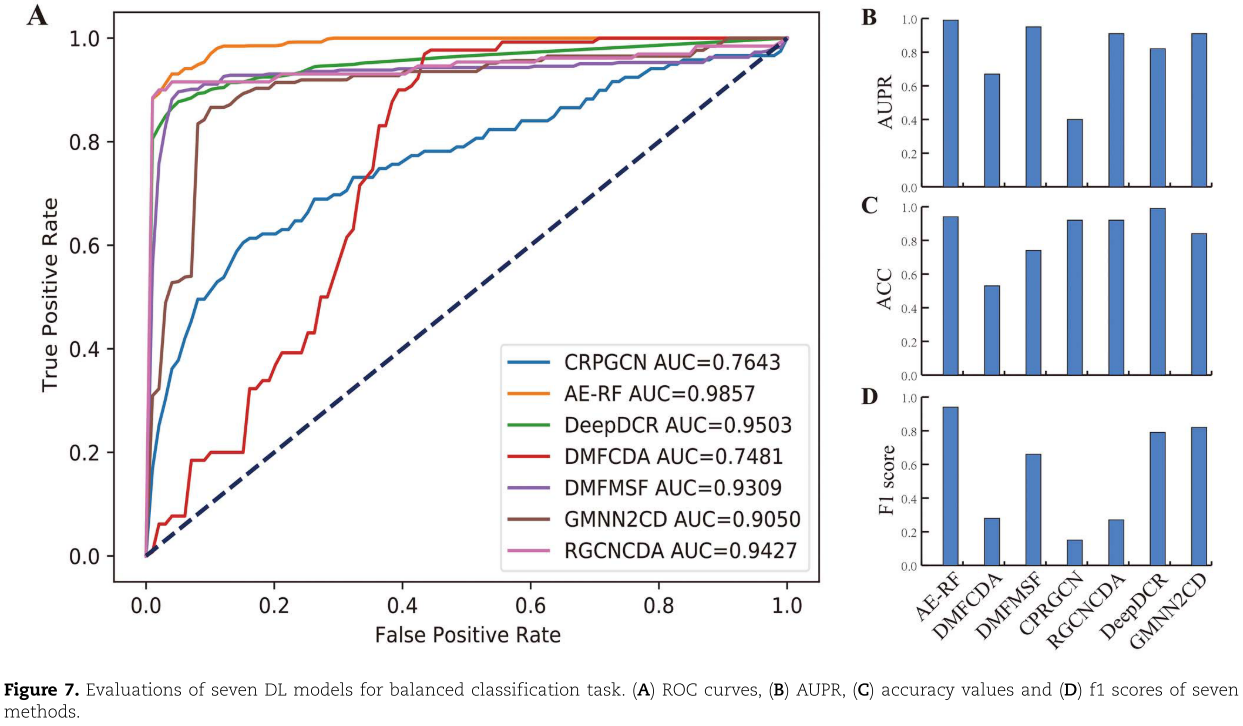
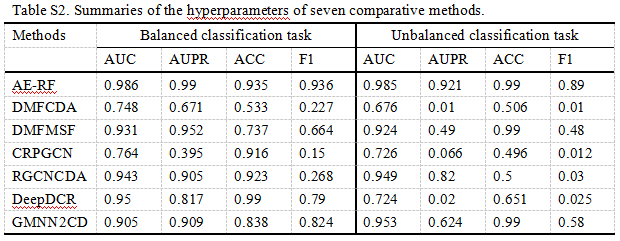
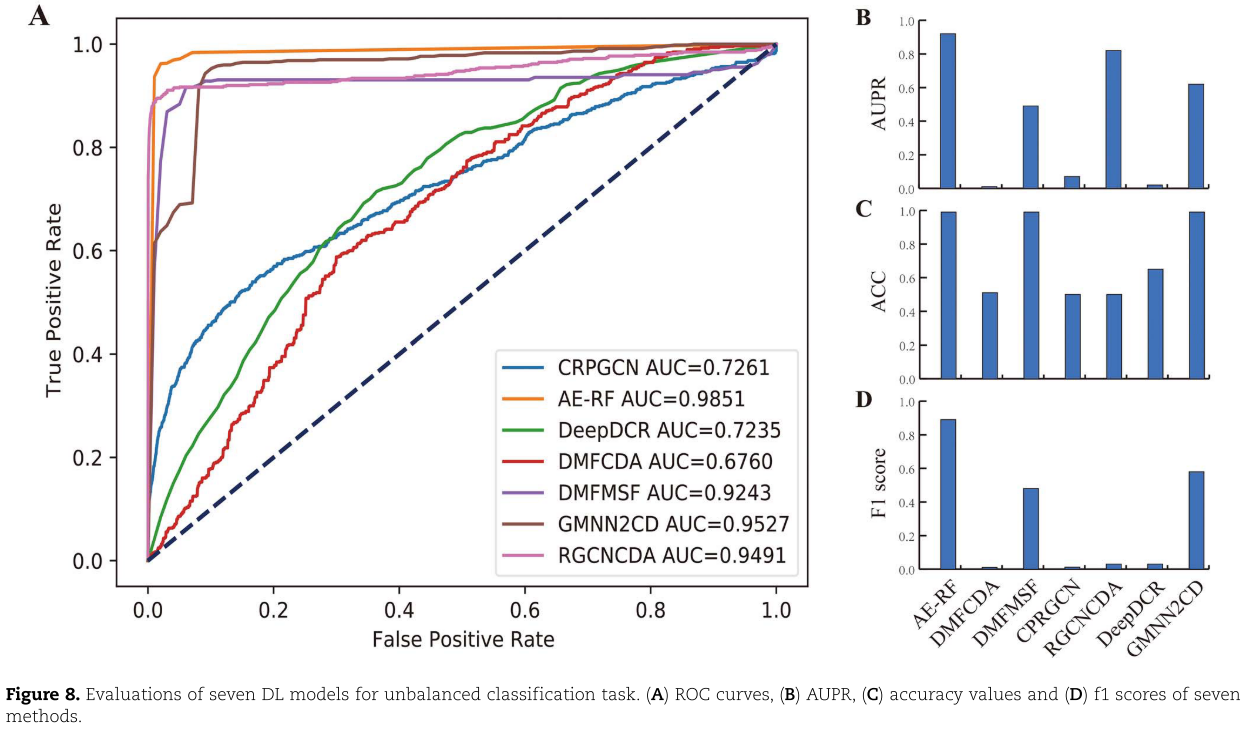
图8总结了七个基于 DL 的非平衡分类任务模型的评估性能。从 ROC 图(图8a)中,我们观察到七种方法之间有很大的不同。其中三个(DMFCDA,CPRGCN,DeepDCR)的表现仅略好于随机预测(AUC = 0.5) ,四个模型,包括 AE-RF,DMFMSF,RGCNCDA 和 GMNN2CD,显示出显着的预测性能,实现中值 AUC 大于0.9。与平衡分类任务相比,只有 DeepDCR 的预测性能急剧下降,而其他六种方法的预测性能相对稳定。对于不平衡的分类任务,AUPR 是一个重要的评估指标,可以更好地反映模型性能的变化(图8B)。七个模型中的三个(DMFCDA,CPRGCN 和 DeepDCR)的中值 AUPR 低于0.1,两个模型(DMFMSF 和 GMNN2CD)具有更好的性能,中值 AUPR 高于0.4但低于0.7。AE-RF 和 RGCNCDA 在 AUPR 方面表现出显著的性能,分别达到0.92和0.82。在 ACC 和 f1评分(图8C 和 D)方面,七个模型中的三个(AE-RF,DMFMSF 和 GMNN2CD)比其他模型具有更好的预测性能。来自七个比较算法的预测结果的具体表现可以在 http:// bib.oxfordjournals.org/的在线补充表 S2中看到。综上所述,评估结果表明,AE-RF、 RGCNCDA 和 GMNN2CD 对于非平衡分类任务具有更显著的性能和更好的相对稳定性。
5.Discussion and conclusion
CircRNA-疾病关联预测研究为疾病发病机制、潜在诊断和治疗靶点的发现提供了深入的见解。此外,DL 承诺为后续的生物实验提供更可靠的候选 circRNAs。本文对14个 circRNA 相关数据库和23种基于 DL 的 circRNA 疾病关联预测方法进行了系统的综述。具体来说,我们将这些方法分为基于特征生成的方法、基于类型鉴别的方法和基于混合的方法(第3节) ,重点介绍了这些方法的核心步骤、深入的模型细节以及各自的优缺点。采用 AUC、 AUPR、 ACC、 f1评分等多项指标,对平衡分类任务和非平衡分类任务的7种代表性方法进行了评价。
值得注意的是,每种方法都有优点和局限性,用户需要根据自己的需求选择合适的方法。对于基于特征生成的方法,关键是通过不同的 DL 模型,如 FastGCN、 GAT、 DeepWalk、 GAN 和 SAE,有效地提取高级特征。基于特征生成的方法的一个优点是自动提取潜在的表征,而不过度依赖于 circRNA 与疾病之间的已知关联。例如,在杨先生的方法中,作者使用 SAE 来自动提取隐藏的深部特征,而不是基于初始相似矩阵的人工选择。基于特征生成方法的另一个优点是实现了多模态特征的互补学习和深度融合,减少了模态间的语义偏差。例如,GATCDA 在图注意机制下工作,通过区分不同邻居的重要性有效地融合多模态特征。然而,大多数基于 DL 模型提取的非线性特征解释能力较差。在基于类型判别的方法中,DL 算法通常用于训练分类器以获得 circRNA-disease 关联分数。一般情况下,基于 DL 的分类器模型具有较高的复杂度,在小数据集训练时容易出现过拟合。例外的是,在 DeepDCR 中,深度森林被用来预测潜在的 circRNA 与疾病的关联。与其他 DL 算法相比,深森林算法需要的超参数少,对小规模数据的处理能力强。在混合方法中,神经网络、深矩阵分解、广义神经网络和广义神经网络同时用于特征生成和类型识别。第一类混合方法结合前两类方法的优点,采用两种不同的 DL 模型分别完成特征生成和类型识别。但是,它们的损失函数需要定制,以便向后传播到全球网络中。第二类基于混合的方法只以端到端的方式使用一个 DL 框架。该模型的一个优点是只有一个目标函数,避免了多模块中某一目标函数与系统宏观目标之间的偏差。另一个优点是它减少了模型的复杂性,一个网络可以处理所有步骤。然而,基于混合的方法很难确定每个模块对最终目标的贡献。此外,当考虑多个不同的任务时,端到端模型不如组装模型灵活。
未来的研究可能集中在两个有希望的方向。一方面,DL 算法在预测 circRNA 与疾病之间的关联方面的作用可以进一步扩大。例如,针对基于特征生成方法可解释性差的缺点,利用深层神经网络对多组学数据进行系统整合和分析,构建客观、有代表性的特征表示方法,使解释结果合理化,从而产生生物学意义。实际上,多组学数据的研究已经成功地应用于 circRNA-疾病关联预测领域。Liu 等[103]利用实体关系网络从收集的 circRNA,药物,lncRNA,miRNA,基因和疾病之间的相互作用数据中挖掘深层规则来预测 circRNA 与疾病之间的关联。这样,就可以从不同的角度描述 circRNAs 的生物学特性和疾病,包括更全面的致病因素。此外,分布式语言算法可以在无监督或无半监督学习的情况下处理极不平衡的分类任务,从而提高预测结果的可靠性。这样做的好处是不需要阴性样品,或者只需要少量的阴性样品。尽管如此,我们仍然缺乏与疾病相关的已知 circRNA,这导致了数据稀疏和限制模型的准确性。因此,与生物学家密切合作收集和积累经过实验验证的生物学数据是一项紧迫的任务。另一方面,本文所研究的 DL 算法在生物信息学的其他领域,如推断药物-疾病关联和药物-靶标相互作用预测等方面也具有潜在的应用前景。例如,深层自动编码器和 DNN 被用来预测癌症的药物敏感性[104,105]。级联深林和 CNN 应用于药物靶标结合亲和力预测[106,107]。进一步,将这些领域与 circRNA 疾病预测相结合,以实现多场景应用。

















 1993
1993

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









