







 本文介绍了如何使用eXpress工具进行转录组和基因组的量化分析。首先,从Gene Expression Omnibus (GEO)和Short Read Archive (SRA)获取数据,然后在没有参考基因组和注释信息的情况下,通过从头组装进行注释。接着,利用Bowtie和Bowtie 2进行比对,并使用eXpress进行靶序列丰度估计。对于已知注释信息的情况,文章也给出了详细的步骤。最后,文章提供了分析results.xprs文件的方法和一些有用的脚本。
本文介绍了如何使用eXpress工具进行转录组和基因组的量化分析。首先,从Gene Expression Omnibus (GEO)和Short Read Archive (SRA)获取数据,然后在没有参考基因组和注释信息的情况下,通过从头组装进行注释。接着,利用Bowtie和Bowtie 2进行比对,并使用eXpress进行靶序列丰度估计。对于已知注释信息的情况,文章也给出了详细的步骤。最后,文章提供了分析results.xprs文件的方法和一些有用的脚本。
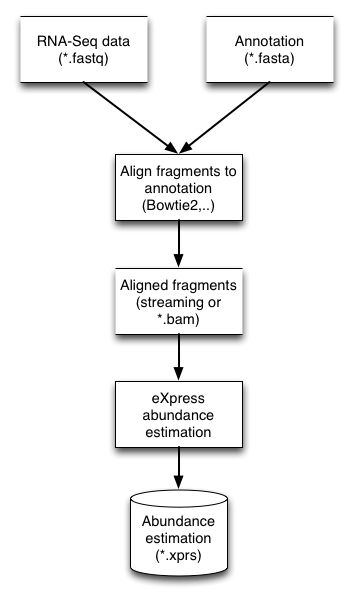
General workflow
eXpress是一个通用的丰度估计工具,它可以应用于任意靶序列和高通量测序reads。 靶序列可以是任意基因组区域,例如RNA-seq中的转录本。因此,一般的流程应该是这样的:
1. 选择你要分析的数据
2. 产生靶序列的集合
3.将目的片段比对到靶序列上
4. eXpress需要的参数包括目的片段,然后进行靶序列的丰度估计
5. 额外的下游分析
图示:
这个教程涉及如下工具:
- Gene Expression Omnibus (GEO) - 取得数据
- The Short Read Archive (SRA) -取得数据
- SRA Toolkit - 从压缩的测序数据中抽取FASTQ文件
- UCSC Genome Browser - 取得注释信息
- Bowtie - 进行比对
- Bowtie 2 -进行比对
- eXpress - 靶序列丰度估计
其它有用的工具(不限于RNA-seq)还包括 here .
例子: 没有参考基因组序列也没有注释信息的情况
有的时候,你将研究没有参考基因组序列的物种,或者参考序列质量较差。这通常意味着你也没有转录组序列。下面要进行的步骤经常是从头组装转录组。接下来,我们将使用Bowtie2进行片段比对。
取得数据
为了取得一个数据,我们将使用GEO访问号。如果你没有一个GEO访问号,而是仅仅想要浏览数据,你可以跟随这个tutorial。为了演示的目的,我们将要研究牦牛转录组。
为什么选择牦牛呢?因为牦牛是天生没有气味的。事实上,是牦牛毛无味。为了下载数据,就直接去GEO吧,然后在"GEO accession" 输入访问号“GSE33300”,点击“GO”。
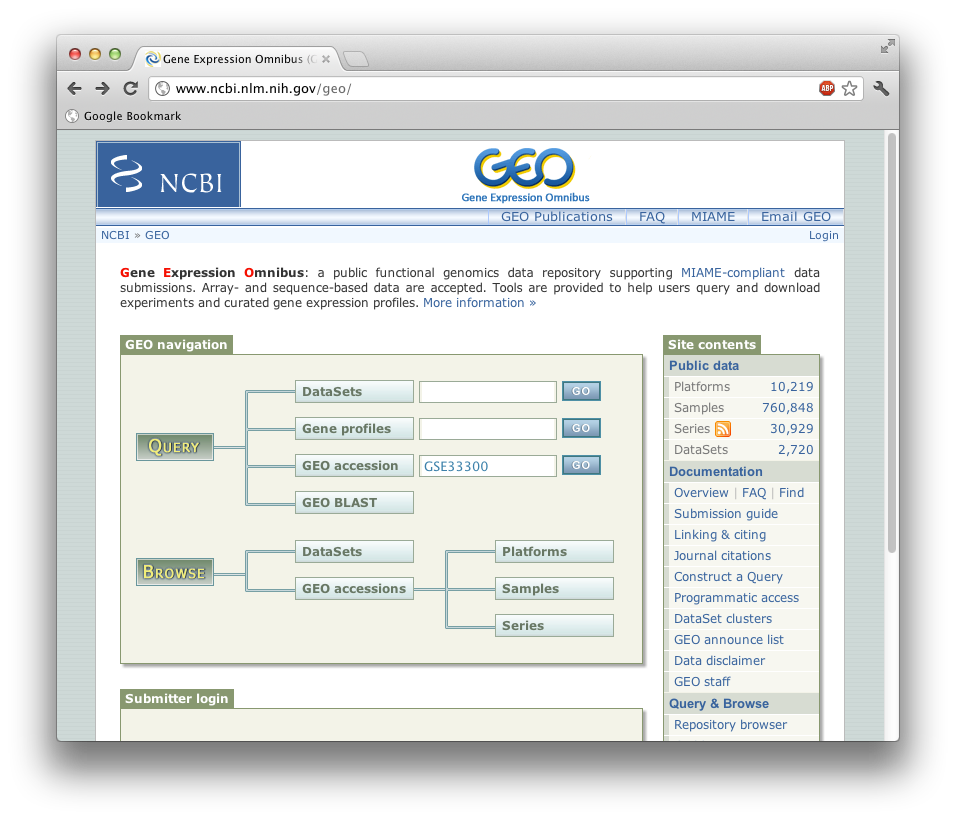
这将将你带到主实验页面。可以看到有6个来自不同器官的不同的样品。我们先看一下"GSM823609 brain"。点击这个实验,并点击ftp链接下载SRA文件。点击目录可以看到SRR361433.sra. 这是一个paired end的RNA-seq数据,我们将使用如下命令抽取数据
- $ fastq-dump --split-3 SRR361433.sra
结果产生两个文件, SRR361433_1.fastq和 SRR361433_2.fastq . 注意到使用 --split-3 . 只有当你下载的数据是paired end的情况下才需要用这个参数.
通过从头组装进行注释
在这个演示中,我们花点时间进行一个完全从头组装分析,而不使用基因组信息,使用的工具是 Trinity .使用如下参数运行Trinity :
- $ Trinity.pl --seqType fq --JM 200G --left SRR361433_1.fastq --right SRR361433_2.fastq --CPU 2
在几个小时内,我们将在trinity_out_dir中得到几个文件 , 包括注释文件 Trinity.fasta .这将是新的注释文件. 从这里开始,如果你有参考基因组的情况下,分析将大致相同(下面的例子也是一样的).
从这里,可以下载组装文件.
比对
建立索引
在你进行任何比对之前,你首先需要建立靶序列的一系列索引文件.- $ cd trinity_out_dir
- $ bowtie2-build --offrate 1 Trinity.fasta Trinity
这将在trinity_out_dir中建立索引,base name是 Trinity,bowtie2需要的参数只需要写到base name结束即可,不需要后面的部分 . 这个索引将允许bowtie2快速将reads比对到靶序列。Offrate 1可以加快比对速度,代价是需要的硬盘空间增大.
进行比对
使用一行命令即可运行bowtie2,- $ bowtie2 -a -X 800 -p 4 -x trinity_out_dir/Trinity \
- -1 SRR361433_1.fastq -2 SRR361433_2.fastq | samtools view -Sb - > hits.bam
- -a - 想让bowtie2报告所有的比对可用此参数,适用于转录组分析,不适用于比对到基因组的分析(eXpress将处理多比对的问题,非常慢!)
- -X 800 - 将设置片段长度(fragment length)不超过800(实际的应用一般设置到300以内就可以了). 这个设置将完全将RNA-seq的测序片段纳入分析的范畴
- -p 4 - 使用4个CPUs进行比对。你应该尽可能多的使用多个CPUs以加快速度.
- -x ... - Bowtie 2索引
- -1 ... -2 ... - RNA-seq实验的左reads和右reads
几十分钟到几小时后(取决于你使用多少CPUs), 你应该看到如下控制台信息:
- [samopen] SAM header is present: 165714 sequences.
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 3万+
3万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









