








从“等价交换”的远古炼金术开始,化学一直是一门了解和控制物质间相互作用的学科。经过不断解锁和利用新的化学反应,人类在研发出新材料便利自己的生活的同时也在提升能量利用效率,促进可持续发展。一个基元化学反应由反应物,过渡态(TS),生成物三者构成。
过渡态是化学中至关重要的 3D 结构,被广泛用于理解化学反应机制、估算反应能垒以及探索庞大的反应网络。然而,由于其在反应过程中存在的时间极短(飞秒量级),实验中几乎不可能分离和表征过渡态。
通常情况下,大家使用量子化学的计算方法来不断求解薛定谔方程,通过已知的反应物和生成物来搜索过渡态。然而,这些计算较为昂贵,并以经常失败而“臭名远扬”。同时,受限于个人的经验直觉和计算所需的资源,每个人所能探索的化学反应也是局限的。这种限制在研究未知的复杂反应时尤为“致命”。它会使研究者忽略到一些潜在可能发生的反应,导致会反应机理的误判,进而影响催化材料设计的思路。
针对这一问题麻省理工学院(MIT)的研究人员开发出了一种基于机器学习的替代方法,可以在几秒钟内发现这些结构。他们的新模型可以帮助化学家探索和设计新的反应和催化剂,以生成诸如燃料化合物或药物之类的高附加值产品,或者模拟自然发生的化学反应,比如推动早期地球生命演化的反应。「过渡态作为设计催化剂或了解自然系统如何执行某些转化的起点,知道其具体结构十分重要」MIT 化学工程和化学教授 Heather Kulik 说道。


论文题目:
Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model
论文链接:
https://www.nature.com/articles/s43588-023-00563-7
相关研究工作以 “Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model” 为题发表在国际顶级期刊 Nature Computational Science 上,其中麻省理工学院的段辰儒博士是第一加通讯作者 [https://rdcu.be/dtGSF]。此外,康奈尔大学博士生杜沅岂,麻省理工学院博士生贾皓钧以及麻省理工学院 Heather Kulik 教授为该论文的共同作者。
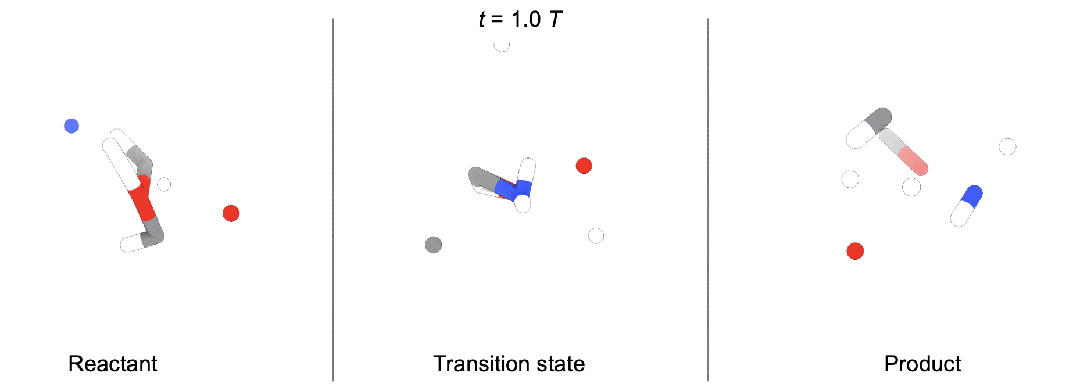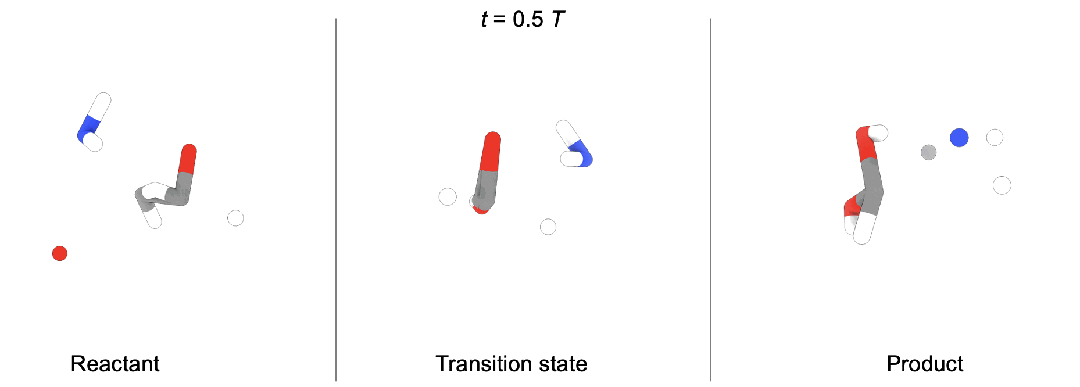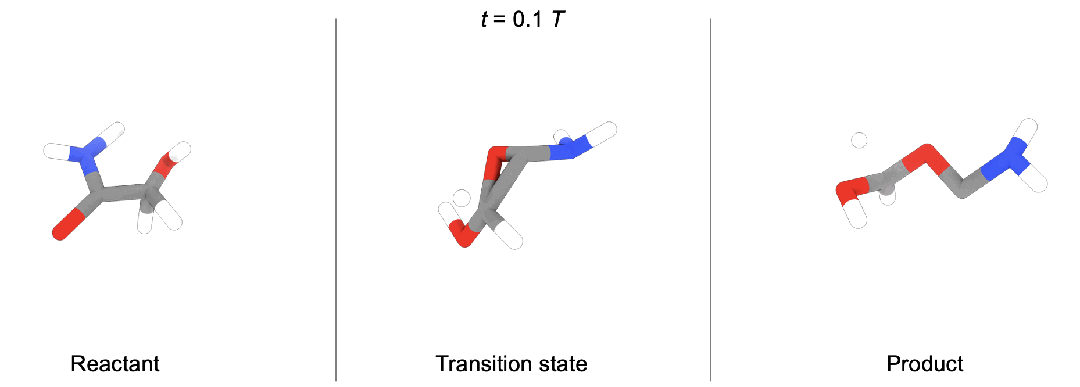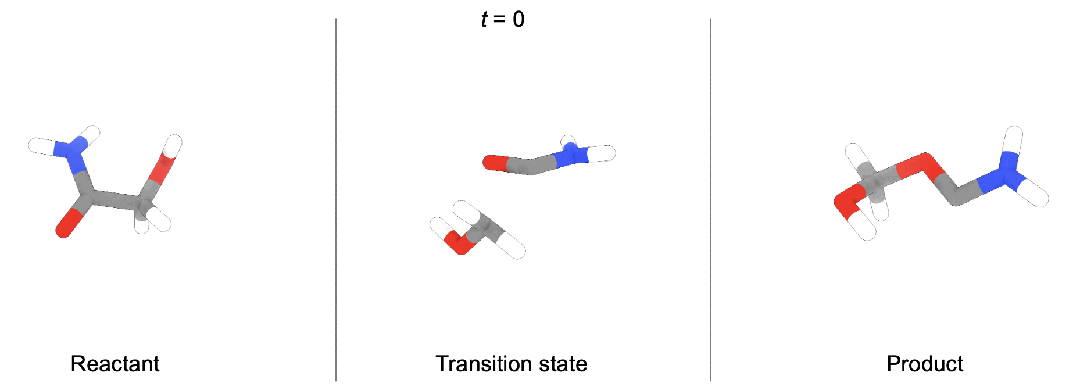

现有问题
1.1 量子化学计算耗时巨大
化学家可以使用一种称为密度泛函理论的量子化学计算方法来计算过渡态。然而,这种方法需要大量的计算资源,需要数小时甚至数天才能完成一个过渡态的计算。
1.2 全连接网络研究化学反应的弊端
为了解决计算时间久的问题,一些研究人员在近期开始尝试使用机器学习模型来发现过渡态结构。然而,迄今为止几乎所有开发的模型都要求将两个反应物建模为一个整体,而反应物之间相对于彼此则保持特定的几何构型(conformation)。任何其他可能的构型都会被机器学习模型误认为成一个新的反应。
「如果反应物分子被旋转,那么从原理上讲,在旋转之前和之后它们仍然可以经历相同的化学反应。就像我们在谈论电解水的时候,只会说水在特定条件下转换为氧气和氢气,而不会描述这些分子的相对几何位置。但在传统的机器学习方法中,模型将把反应物和生成物在不同几何位置的反应视为两个不同的反应。这使得机器学习训练变得更加困难,准确性也会随之下降」段辰儒博士表示。
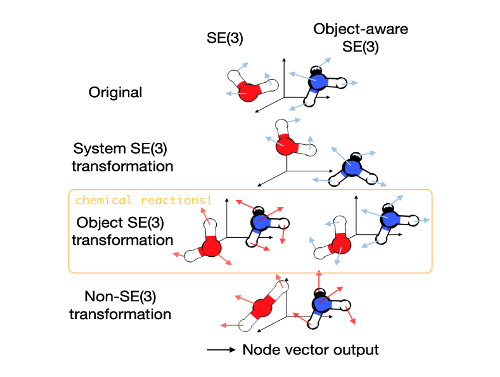
1.3 扩散模型的局限
扩散模型作为一个生成式模型曾被应用于图像处理中。最近,扩散模型还被应用于 3D 分子和蛋白结构生成、蛋白质-配体对接以及基于结构的药物设计。在这些应用中,扩散模型利用 3D special Euclidean group (SE(3)) 图神经网络(GNNs)来保留分子的排列、平移和旋转对称性。
然而,由反应物、过渡态和产物组成的基元反应遵循“对象感知“的 SE(3) 对称性。这是因为基元反应中三个对象之间的相互作用不是通过 3D 欧几里得空间进行的,而是在更高维的电子势能面(potential energy surface)的因果联系。因此,现有的基于 SE(3) GNN 的扩散模型会因为破坏对称性而存在问题。

解决方案
2.1 扩散模型描述化学反应过程
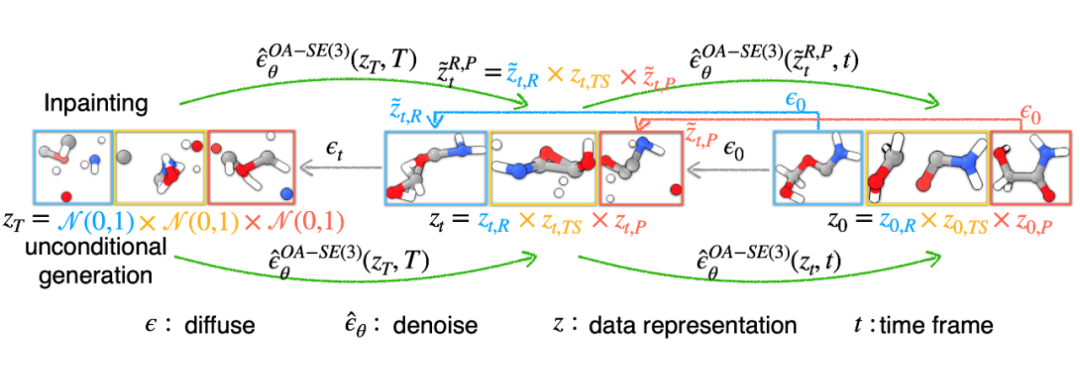
段辰儒博士等人首次将扩散生成推广到多体系生成。训练时,同时向「反应物(蓝),过渡态(黄),生成物(红)」加入噪声,并使用一个拥有特殊对称性(后面会详细描述)的图神经网络模型预测加入的三个噪声。生成时,他们开发了两种模式。
随机生成。这种情况下,直接从三个高斯分布出发,生成新的化学反应,即全新未知的「反应物,过渡态,生成物」。
条件生成。在化学反应中,通常大家知道部分信息。段辰儒博士在这里使用内绘制(inpainting)条件生成。比如给定反应物和生成物,直接生成过渡态。这个过程中,每一步都将「反应物, 生成物」中加入固定噪声,并将它们与图神经网络模型预测得到的过渡态结合。一步步迭代,直到产生「反应物(“已知”),过渡态(“未知”),生成物(“已知”)」。
2.2 等变扩散模型的建立
扩散模型的灵感最初来自非平衡热力学。一个扩散模型有两个过程,即正向(扩散)过程和反向(去噪)。其中正向过程是在数据中逐渐添加噪声,直到噪声成为高斯分布。
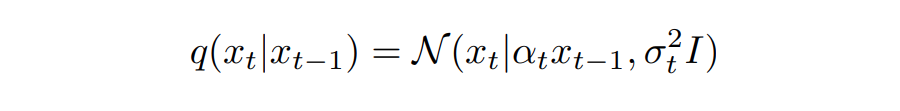
而其中的 用来控制保留的信号, 用来控制增加的噪声。信噪比也被定义为 SNR(t)=,其中。由于高斯噪声的特性,真正的去噪过程可以用闭合形式写出
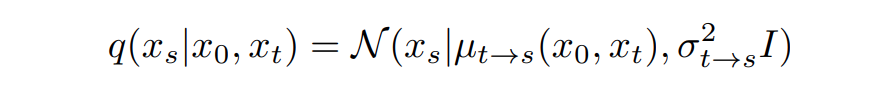
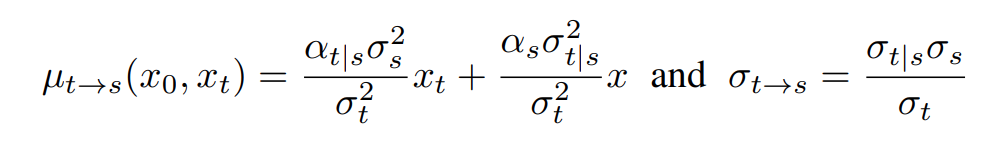
其中, 指的是扩散/变色过程中从 0 到 T 的两个不同时间,。然而,这个真正的去噪过程是依赖于 的。但 是数据分布,理论上无法获取。因此,扩散学习的去噪过程是用去噪网络 预测的 替换 。训练目标是最大化训练数据似然值的变异下限(VLB)。
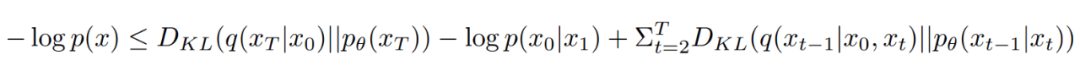
要建立一个 SE(3)-equivariant 扩散模型,需要一个 SE(3)-invariant 先验的核和一个 SE(3)-equivariant 过渡的核。为了保证在排列、旋转和平移时的不变性,一个必要条件就是使用 SE(3)-equivariant 过渡核(即去噪网络)。其中对旋转和平移的性质还有额外的要求。对于旋转而言,各向同性高斯先验具有很好的等变量变换特性。
2.3 对象感知 SE(3) 的实现
一般来说,由多个对象(分子或蛋白质)组成的系统,其对象在 3D 欧几里得空间中没有相互作用,可以用一组独立的图 来描述。举一个例子,在基本反应中有三个可以索引的对象,反应物(i=0),过渡态(i=1)和产物(i=2)。对于这些系统的一个重要的对称性是 SE(3) 等变性。这意味着在系统中对每个单独的对象进行任何 SE(3) 变换都不应影响其描述:
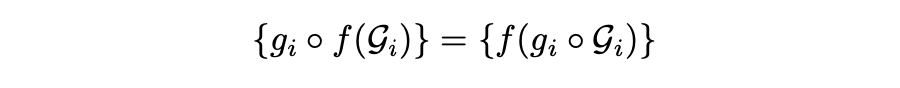
表示对第 个对象进行的 SE(3) 变换,这并不一定对所有对象都相同。另一方面,在系统中对任何对象进行的非 SE(3) 过渡都应该对其描述产生下面所展示的影响:
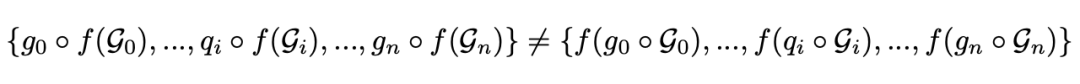
其中, 表示对第 个对象进行的非 SE(3) 变换。SE(3) GNN 在这一情况下适用于后者,但是会违反前者的对称性。
为了同时满足这两个要求(以及 SE(3) 等变扩散模型自然满足的其他对称性),段辰儒博士等人开发了一种通用方法,使任何 SE(3) GNN 能够适应其对象感知的 SE(3) 等效形式。
该方法的核心思想是仍然使用 SE(3) GNN 对单个对象执行等变更新,但仅允许在不同对象之间进行标量类型的消息传递,以避免它们的相对位置信息的“泄漏”。从原始图表示 开始, 首先通过一个 SE(3) 等变块进行消息传递的更新。将所有对象的结果图()标量化并连接成一个系统级的全连接图。
然后,通过一个标量消息传递块更新 中的所有标量节点特征。通过这种方式,系统中不同对象(i)之间的相互作用也会被包含在内,但不会引入不同对象的定位,因为所有高阶张量都已被标量化。最后,更新后的节点标量与等变更新块的输出相结合,构成了建立在基本 SE(3) 更新函数之上的对象感知 SE(3) 交互块。该交互块与SE(3) GNNs类似,将通过多次重复最终实现图的读取。
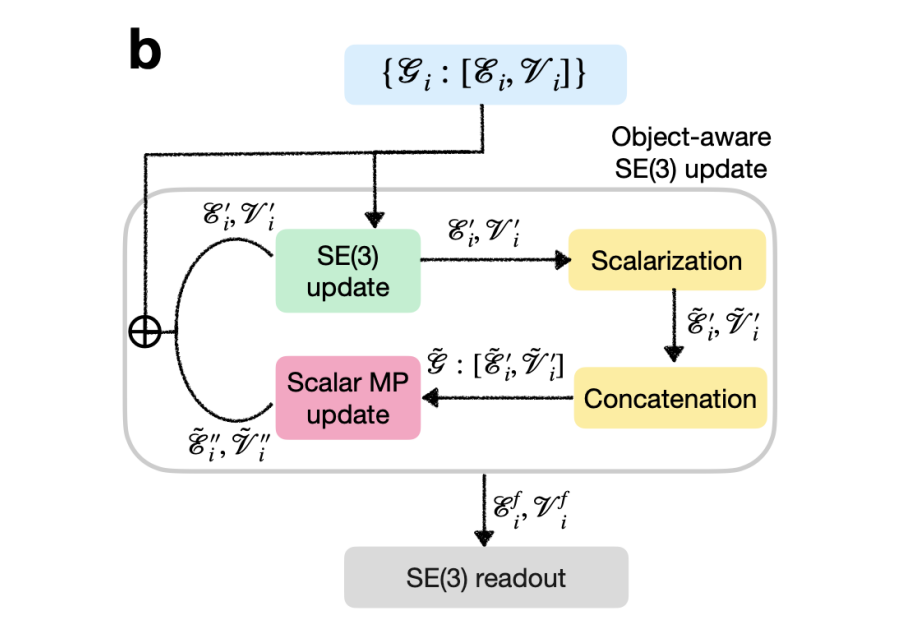
▲ 图1. “对象感知“的SE(3) GNN实现方法

总体性能
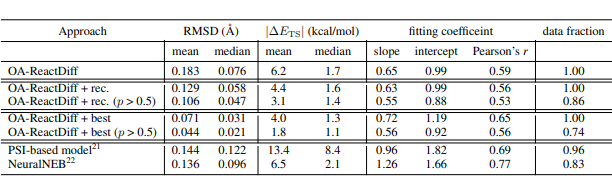
▲ 表1. TS 结构的 RMSD 和绝对能量差异的统计摘要以及各种方法获得的线性拟合结果。
在训练集中,研究人员使用量子计算方法得到了 9,000 种不同化学反应的反应物、过渡态和生成物结构。并在约 1,000 个之前未见过的反应上进行了测试,要求它为每个过渡态生成 40 种可能的结构。在计算的过程中通过引入“推荐模型”来预测哪个过渡态的置信度最高。在此基础上进一步结合不确定性估计,研究人员在仅对 14% 模型不确定性最高的反应执行量子化学计算,就实现了 2.6 kcal/mol 的平均绝对误差。
这使得在使用 OA-ReactDiff 估算 300°C 的反应速率时,可以得到一个数量级误差范围内的结果。OA-ReactDiff 生成的结构与量子化学计算得到的过渡态结构相比,均方根误差(RMSD)在 0.06 埃(千分之六纳米)范围内,这个误差量级在肉眼种几乎不可区分。更令人欣喜的是 OA-ReactDiff 生成一个过渡态结构只需要 6 秒,相比于量子化学计算至少加速了 1000 倍。由此,该算法成功实现了对 TS 结构和反应能垒计算的极高准确性和快速性。
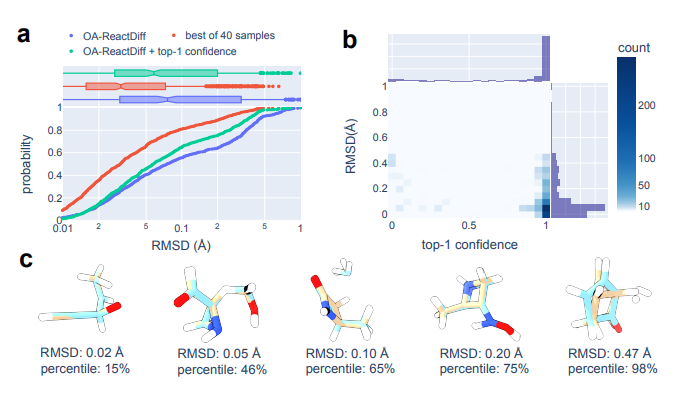
▲ 图2. 通过 OA-ReactDiff 生成的 TS 结构与真实 TS 结构的结构相似性评估。
针对 OA-ReactDiff 和 Transition1x 中 1,073 个未见反应的真实 TS 结构之间的结构相似性,团队以 RMSD 位标准做出评估。与比较原子局部几何形状的键长、角度和二面体相比,RMSD 应该提供对整体结构一致性的更准确评估,这是 TS 搜索的最终目标。
对于每个反应,团队运行了 40 次 OA-ReactDiff,生成了 40 个独立的 TS 结构猜测。40 个随机选择的样本中,1073 个测试基本反应里,OA-ReactDiff 的平均 RMSD 值已达到 0.183 Å,中值为 0.076 Å(图2a)。
此外,推荐的结构主要位于低 RMSD 和高置信度区域,这证明了综合使用 OA-ReactDiff 和置信度推荐方法的有效性(图2b)。RMSD 误差的平均值和中值分别为 0.129 和 0.058 Å,大约三分之二的推荐 TS 结构的 RMSD 值 < 0.1 Å(图2a)。
团队同样观察到,随着独立运行总数的增加,OA-ReactDiff + 推荐器方法的性能也在不断提高。此外,团队对每个样本进行了 40 次运行,以在总运行时间(总共 4 分钟)和采样精度之间取得平衡。尽管推荐器在区分低 RMSD(即,< 0.2 Å)的结构方面还远远不够完美,但它有助于避免与真实 TS 结构非常不同的 TS 样本(即,> 0.45 Å,图2c)。
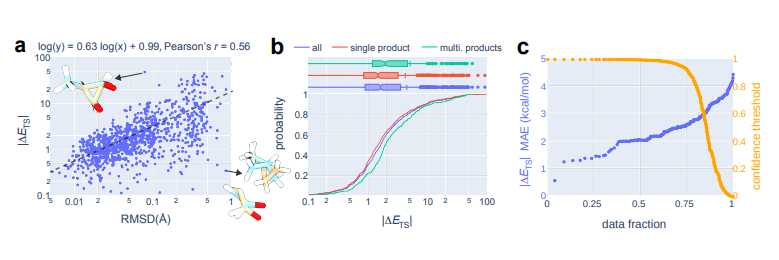
▲ 图3. OA-ReactDiff + 推荐器TS结构的能量性能
除了提供微观层面的基本反应如何发生的确切 3D 结构外,TS 搜索算法还应评估反应势垒高度,这对于精简大型反应网络和估算反应速率至关重要。
团队比较了 OA-ReactDiff 推荐的 TS 结构和真实 TS 结构之间的电子能量以评估势垒高度。正如人们可能期望的那样,OA-ReactDiff 和真实 TS 结构之间的绝对能量差()与它们的 RMSD 呈正相关关系。
团队发现它们在各种常见代数拟合类型上都遵循幂的规律,即在 log-log 图上给出了 0.56 的 Pearson 相关系数(图 3a)。这相对较低的 Pearson 相关系数可以解释为分子的势能面的高复杂性,其中平移一个原子的方向对能量变化有很大的影响。
例如,对于 的一个 OA-ReactDiff TS,与真实 TS 相比的 RMSD 值相对较低,为 0.078 Å,但在 1073 个测试反应中,其能量差异最大(49.3 kcal/mol)(图 3a)。
另一方面,尽管 OA-ReactDiff 和 的真实 TS 结构之间的 RMSD 极大(0.821 Å),但两种结构之间的能量差仅为 0.4 kcal/mol,其原因便是该 TS 由两个仅弱相互作用的片段组成(图 13)。
而使用 OA-ReactDiff 和推荐器,团队得到了生成的 TS 结构与真实 TS 结构之间的绝对能量差的平均值为 4.4 kcal/mol,中值为 1.6 kcal/mol,其中 71% 的 TS 势垒误差 < 3 kcal/mol。
此外,团队观察到对于 TS 结构生成的置信度阈值的 MAE 和置信度阈值之间存在单调关系(图 3c)。这种被期望的单调行为表明,大家可以利用置信度分数进行不确定性量化,以在将 OA-ReactDiff、推荐器和基于 DFT 的 TS 搜索结合的实际工作流程中平衡准确性和所需的 DFT 计算数量。
3.1 局限性与未来期许
这项工作是 3D 扩散模型首次在化学反应中的亮相。尽管研究人员主要仅对较小数量原子的化合物(<25 个原子)的反应上进行了模拟训练,但他们发现整个模型也能够对较大分子的反应进行准确预测。Kulik 教授说:「即使你面对更大的系统甚至是酶催化的系统,你仍然可以得到关于原子最有可能重新排列的不同方式」。
研究人员现在计划加入其他组分来扩展他们的模型,比如催化剂。借助生成式AI的随机性,OA-ReactDiff 可以探索到意料之外的化学反应。这个特点弥补了现有基于化学的直觉反应探索框架,帮助建立更加完整的化学反应网络,助力研发设计新型催化材料。
这方面的研究可以帮助他们加速发掘对特定反应的新的催化剂。此外,他们提出的算法对于开发药品、燃料或其他有用化合物的新过程可能非常有用,尤其是当合成涉及许多化学步骤时。「传统上,所有这些计算都是用量子化学进行的,而现在我们能够用更快的生成模型替代量子化学」段辰儒博士说。贾皓钧也表示「化学反应是化学研究的核心,但人类历史上大部分重要的化学反应都是偶然发现的。
生成模型的创造性可以帮助探索传统筛选中被忽略的化学反应,为设计新型催化材料和解锁新的化学反应开辟了全新的途径,不断解锁新的化学反应为人类创造更美好的未来」。除了催化剂设计这种偏“工业型“的可能,OA-ReactDiff 还有许多有意思的潜在应用,如探索可能发生在其他行星上的气体间相互作用,模拟地球早期生命演化过程中发生的反应过程等等。
文章链接:
C. Duan*, Y. Du, H. Jia, and H. J. Kulik, “Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model”, Nat. Comput. Sci., ASAP,
https://www.nature.com/articles/s43588-023-00563-7
更多阅读




#投 稿 通 道#
让你的文字被更多人看到
如何才能让更多的优质内容以更短路径到达读者群体,缩短读者寻找优质内容的成本呢?答案就是:你不认识的人。
总有一些你不认识的人,知道你想知道的东西。PaperWeekly 或许可以成为一座桥梁,促使不同背景、不同方向的学者和学术灵感相互碰撞,迸发出更多的可能性。
PaperWeekly 鼓励高校实验室或个人,在我们的平台上分享各类优质内容,可以是最新论文解读,也可以是学术热点剖析、科研心得或竞赛经验讲解等。我们的目的只有一个,让知识真正流动起来。
📝 稿件基本要求:
• 文章确系个人原创作品,未曾在公开渠道发表,如为其他平台已发表或待发表的文章,请明确标注
• 稿件建议以 markdown 格式撰写,文中配图以附件形式发送,要求图片清晰,无版权问题
• PaperWeekly 尊重原作者署名权,并将为每篇被采纳的原创首发稿件,提供业内具有竞争力稿酬,具体依据文章阅读量和文章质量阶梯制结算
📬 投稿通道:
• 投稿邮箱:hr@paperweekly.site
• 来稿请备注即时联系方式(微信),以便我们在稿件选用的第一时间联系作者
• 您也可以直接添加小编微信(pwbot02)快速投稿,备注:姓名-投稿

△长按添加PaperWeekly小编
🔍
现在,在「知乎」也能找到我们了
进入知乎首页搜索「PaperWeekly」
点击「关注」订阅我们的专栏吧
·
·


















 2242
2242

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









