






平台:Ubuntu Linux平台上进行。
在进行16S扩增子实验前,我们需要安装好fastqc,相关教程在我的其他文档,如:在Linux中的qiime2。安装一天的fastqc,找到最好的解决办法-CSDN博客
一、文件要求:1、fq,fastqc等格式。
2、mapping.txt。linux中的示例:

3、manifest.txt,linux中示例:
以上文件存放在同一个文件夹里并命名为dna_data.
二、1、激活qiime2。conda activate qiime2-amplicon-2023.9#(qiime2-amplicon-2023.9为你下的版本)
2、在qiime2的根目录中开始进行命令操作。
3、 输入文件 user@user:~$ qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path /home/user/dna_data/manifest.txt \
--output-path result/paired-end-demux.qza \
--input-format PairedEndFastqManifestPhred33
(/home/user/dna_data/manifest.txt 绝对路径可以防止报错,下文输入的路径全是绝对路径)
成功标志:Imported /home/user/dna_data/manifest.txt as PairedEndFastqManifestPhred33 to result/paired-end-demux.qza
三、以下操作都在cd result的文件下进行操作
1、 去除引物序列(本人的数据不知道引物序列,因此没做)
引用原话:
本测序数据的引物为338F和806R, p-front-f和p-front-r处分别输入自己的F和R端引物序列。
nohup time qiime cutadapt trim-paired \
--i-demultiplexed-sequences paired-end-demux.qza \
--p-front-f ACTCCTACGGGAGGCAGCA \
--p-front-r GGACTACHVGGGTWTCTAAT \
--o-trimmed-sequences paired-demux.qza2、创建可视化文件查看数据
user@user:~/result$ qiime demux summarize \
--i-data paired-end-demux.qza \
--o-visualization demux.qzv
成功标志:Saved Visualization to: demux.qza.qzv
3、data2降噪、序列质量控制、构建特征表和代表序列表
人家老大哥原话,更好理解:在qiime2网站中查看上一步得到的paired-demux.qzv,得到碱基质量值下降位点,使用p-trunc-len-f和p-trunc-len-f切除质量差的碱基。
user@user:~/result$ time qiime dada2 denoise-paired \
--i-demultiplexed-seqs paired-end-demux.qza \
--p-trunc-len-f 228 \
--p-trunc-len-r 215 \
--o-table table.qza \
--o-representative-sequences rep-seqs.qza \
--o-denoising-stats denoising-stats.qza
成功标志:Saved FeatureTable[Frequency] to: table.qza
Saved FeatureData[Sequence] to: rep-seqs.qza
Saved SampleData[DADA2Stats] to: denoising-stats.qza
4、查看去噪过程统计
user@user:~/result$ qiime metadata tabulate \
--m-input-file denoising-stats.qza \
--o-visualization denoising-stats.qzv
成功标志:Saved Visualization to: denoising-stats.qzv
5、查看统计特征表
user@user:~/result$ qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file /home/user/dna_data/mapping.txt
(/home/user/dna_data/mapping.txt。绝对路径)
成功标志:Saved Visualization to: table.qz
6、①.导出feature table
user@user:~/result$ qiime tools export --input-path table.qza \
--output-path feature_table
成功标志:Exported table.qza as BIOMV210DirFmt to directory feature_table
user@user:~/result$ biom convert -i feature_table/feature-table.biom \
-o feature_table/feature-table.txt --to-tsv
示例: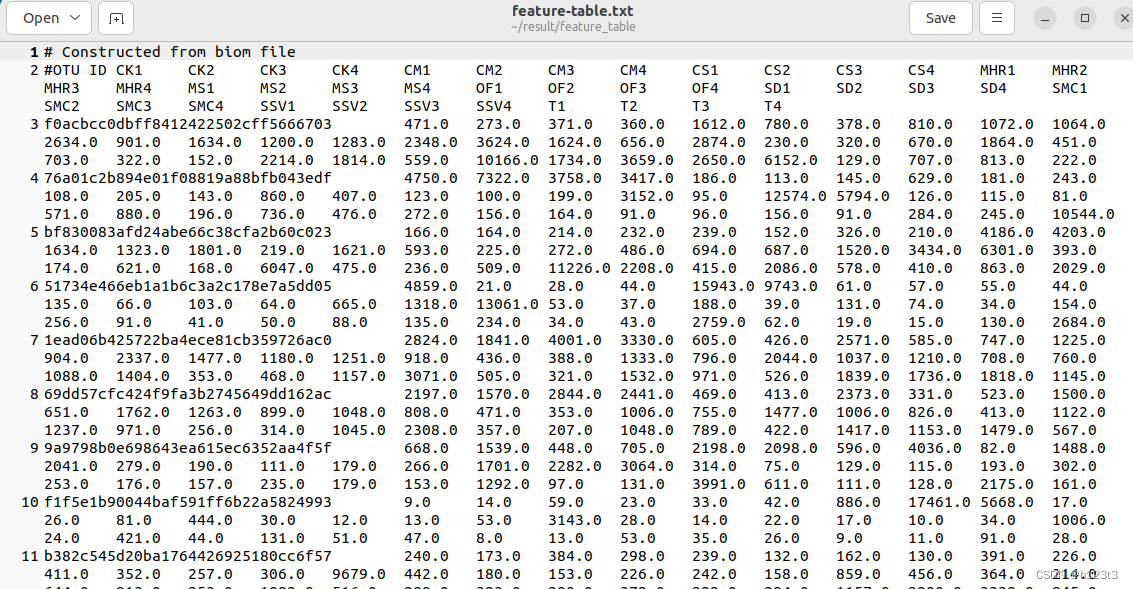
②. 计算relative frequency,并导出feature table
相关性: ser@user:~/result$ qiime feature-table relative-frequency --i-table table.qza \
--o-relative-frequency-table table-relative.qza
成功标志:Saved FeatureTable[RelativeFrequency] to: table-relative.qza
导出feature table:user@user:~/result$ qiime tools export --input-path table-relative.qza \
--output-path feature_table_relative
成功标志:Exported table-relative.qza as BIOMV210DirFmt to directory feature_table_relative
user@user:~/result$ biom convert -i feature_table_relative/feature-table.biom \
-o feature_table_relative/feature-table_relative.txt --to-tsv
示例:
为了以防以后忘记物种注释,本人将前人的记录摘抄下来,如下:
下载已建好的silva分类器:
由于训练Silva分类器需要花费大量时间,而Qiime2官网有已训练好的分类器,我们就用现成的了。本文使用的分类器为Silva 138按99%相似度聚类OTUs的全长序列。如果测序数据的引物序列为515F和806R,可以选择Silva 138按99% OTUs聚类V4区515F/806R的序列。
wget -c https://data.qiime2.org/2023.9/common/silva-138-99-nb-classifier.qza生成物种注释表
nohup time qiime feature-classifier classify-sklearn \
--i-classifier silva-138-99-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza &可视化物种注释表
time qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv














 760
760











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









