







目录
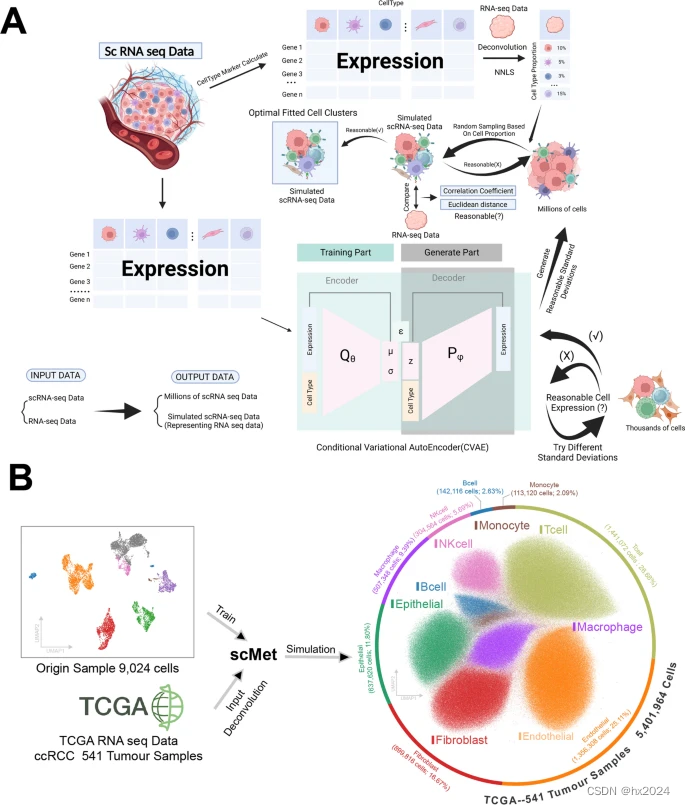
收集透明细胞肾细胞癌9个主要单细胞RNA测序数据库,包含195个样本。选取空间转录组学资料进行空间定位代谢活性分析。开发scMet程序,将RNA-seq数据转换为scRNA-seq数据,用于下游分析。
肿瘤微环境异质性
收集了透明细胞肾细胞癌(ccRCC)的公开单细胞RNA测序数据,并进行了质量控制和过滤。最后,我们保留了来自 76 名患者的 195 份样本,用于肿瘤微环境的高分辨率定位,包括来自 981,294 个细胞的转录组数据,用于后续分析
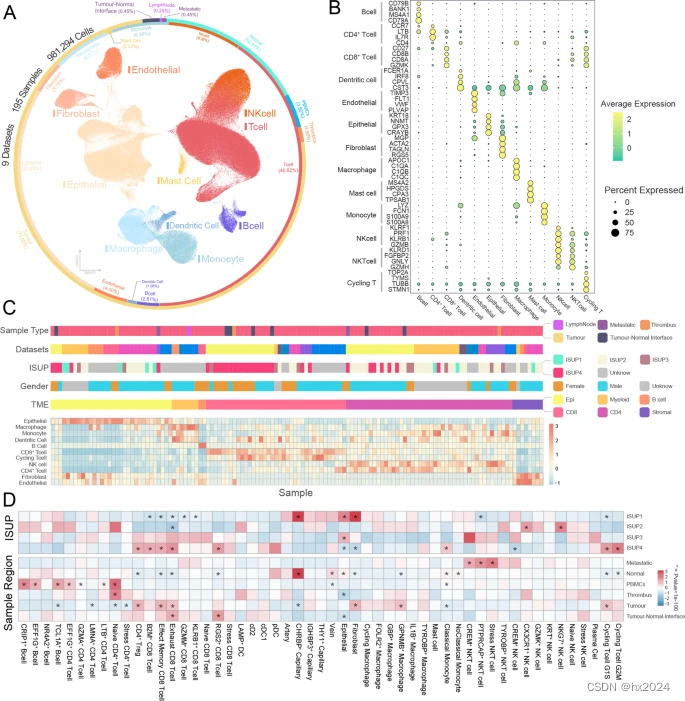
透明细胞肾细胞癌的高分辨率单细胞图谱。ccRCC 的 UMAP 可视化,内环表示细胞类型比例,外环表示组织来源比例。B 点图说明了主要细胞类型的标记基因表达。C 显示样本信息、肿瘤微环境分类组和肿瘤样本的细胞浸润富集水平的热图。D 热图显示了不同 ISUP 组织学等级和采样点的肿瘤微环境中的细胞浸润富集水平。ISUP:国际泌尿病理学会;UMAP:均匀流形近似和投影.
肿瘤细胞和正常细胞之间代谢差异
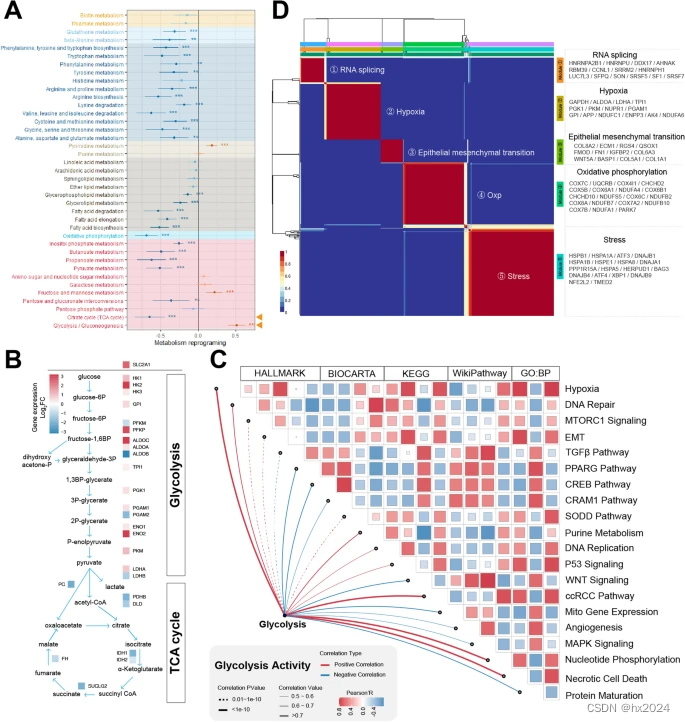
透明细胞肾细胞癌肿瘤细胞的糖酵解活性明显增强。描绘肿瘤细胞和正常上皮细胞之间多种代谢途径差异活性的森林图,不同的背景颜色代表不同的代谢类别。B 肿瘤细胞和正常上皮细胞之间糖酵解和三羧酸 (TCA) 循环相关基因的差异表达,红色突出显示在肿瘤细胞中高表达,蓝色表示在正常上皮细胞中高表达。C 显示肿瘤细胞中糖酵解活性与各种生物学功能之间关联的相关热图。D 肿瘤细胞不同状态的相关性热图,其中红色表示高相关性,蓝色表示低相关性。EMT:上皮-间充质转化;Oxp:氧化磷酸化
CD8 T细胞免疫应答中与糖酵解
使用完善的谱系标记,我们将 T 细胞进一步分为不同的亚型:CD8 T 细胞 (45.52%,CD8A/CD8B/GZMM)、CD4 T 细胞 (18.50%,IL7R/CD4)、NKT 细胞 (8.70%,CD3E/GZMH)、CD4 Tregs (5.41%,FOXP3/TIGIT/CTLA4)、循环 T 细胞 (5.41%,MKI67/TOP2A) 以及 NK 细胞 (16.55%,NKG7/GNLY/KLRD1)
 肿瘤微环境中T细胞的代谢异质性和动力学。T 细胞和 NK 细胞的 UMAP 图,按细胞类型进行颜色编码。B 肿瘤微环境和正常组织微环境之间各种 T 细胞亚型代谢活性的差异,由实心圆圈和空心圆圈表示,表示 p 值分别< 0.05 和 > 0.05。C 各种T细胞类型中糖酵解活性与多种生物学功能的相关性。CD8 T 细胞的 D UMAP 图,按细胞亚型进行颜色编码。CD8 T 细胞的 E UMAP 图,通过推断的伪时间轨迹位置进行颜色编码,范围从黑色(轨迹起点)到黄色(轨迹终点)。F 沿推断的假时间轨迹在T细胞中的耗尽标记基因表达。G 线图描绘了沿推断的伪时间轨迹的细胞毒性和幼稚 T 细胞标志基因的表达。H线图显示了能量代谢活动沿推断的伪时间轨迹的动态变化。I 折线图说明了氨基酸代谢活性沿推断的伪时间轨迹的动态变化。J CD8 T细胞转录因子活性热图,红色表示高活性,蓝色表示低活性。UMAP:均匀流形近似和投影+++。
肿瘤微环境中T细胞的代谢异质性和动力学。T 细胞和 NK 细胞的 UMAP 图,按细胞类型进行颜色编码。B 肿瘤微环境和正常组织微环境之间各种 T 细胞亚型代谢活性的差异,由实心圆圈和空心圆圈表示,表示 p 值分别< 0.05 和 > 0.05。C 各种T细胞类型中糖酵解活性与多种生物学功能的相关性。CD8 T 细胞的 D UMAP 图,按细胞亚型进行颜色编码。CD8 T 细胞的 E UMAP 图,通过推断的伪时间轨迹位置进行颜色编码,范围从黑色(轨迹起点)到黄色(轨迹终点)。F 沿推断的假时间轨迹在T细胞中的耗尽标记基因表达。G 线图描绘了沿推断的伪时间轨迹的细胞毒性和幼稚 T 细胞标志基因的表达。H线图显示了能量代谢活动沿推断的伪时间轨迹的动态变化。I 折线图说明了氨基酸代谢活性沿推断的伪时间轨迹的动态变化。J CD8 T细胞转录因子活性热图,红色表示高活性,蓝色表示低活性。UMAP:均匀流形近似和投影+++。
代谢异质性塑造肿瘤微环境中的巨噬细胞功能
但其在ccRCC中的表现尚未阐明。为了进一步分析巨噬细胞的代谢差异,我们分离了巨噬细胞(C1QA/C1QB/C1QC),进行了UMAP聚类,并根据高表达基因对簇进行了命名。簇分别为IL1B巨噬细胞(26.20%,IL1B/EREG/AREG)、FOLR2巨噬细胞(23.50%,FOLR2/EGR1/MAF)、GBP巨噬细胞(16.78%,GBP1/GBP4)、GPNMB巨噬细胞(15.27%,GPNMB/APOC1/CTSD)、S100A8巨噬细胞(11.87%,S100A8/FTL)和循环巨噬细胞(6.40%,MKI67/TOP2A)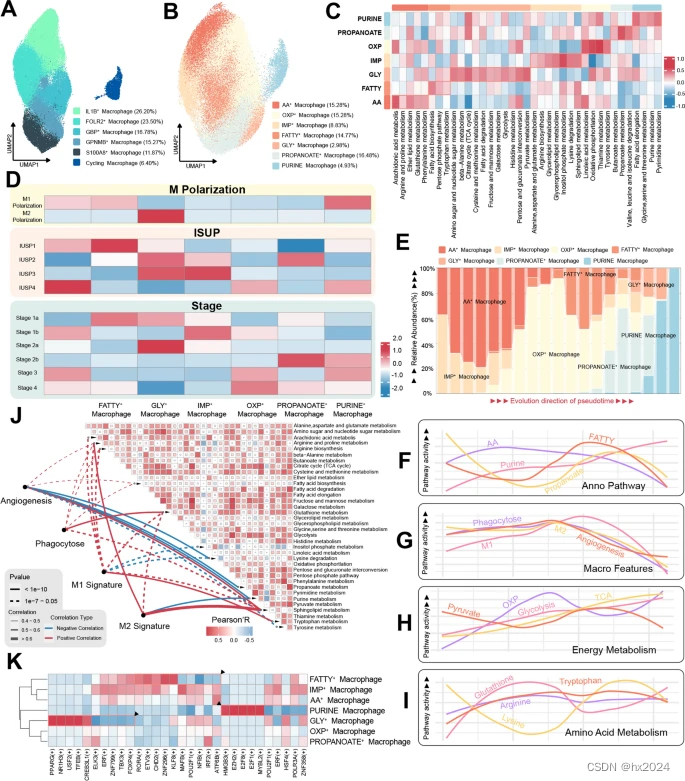
巨噬细胞代谢及其高度相关的生物学意义。巨噬细胞的UMAP图,按细胞亚型进行颜色编码。B 巨噬细胞的UMAP图,按代谢亚型进行颜色编码。C 不同代谢亚型巨噬细胞的代谢活性热图。D 不同极化、ISUP 组织学分级和肿瘤分级中巨噬细胞亚型的浸润富集热图。E 不同巨噬细胞亚型富集水平沿推断的假时间轨迹的动态变化。F 巨噬细胞代谢亚型注释代谢通路活性沿推断的假时间轨迹的动态变化。G 巨噬细胞特征活性沿推断的伪时间轨迹的动态变化。H 巨噬细胞能量代谢活性沿推断的假时间轨迹的动态变化。I 巨噬细胞氨基酸代谢活性沿推断的假时间轨迹的动态变化。J 巨噬细胞特征活性与各种代谢途径活性之间的相关热图。K 巨噬细胞转录因子活性热图,其中红色表示高活性,蓝色表示低活性。IMP:肌醇磷酸代谢;AA:花生四烯酸代谢;OXP:氧化磷酸化;GLY:糖酵解;脂肪:脂肪酸代谢;ISUP: International Society of Urological Pathology.UMAP: 均匀流形近似和投影
ENPP2潜在标志物
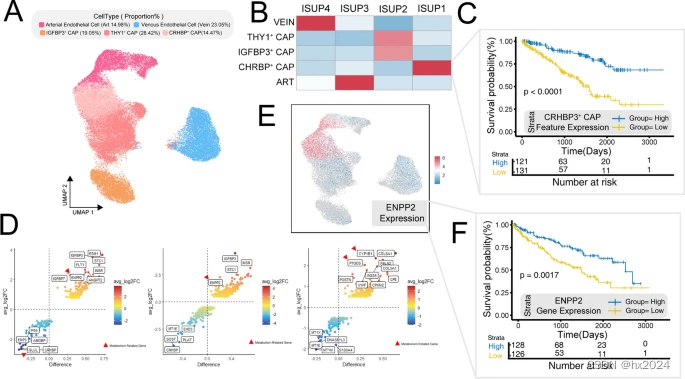
内皮细胞表达异质性。内皮细胞的 UMAP 图,按细胞亚型进行颜色编码。B 不同 ISUP 等级的肿瘤组织中不同内皮细胞亚型的富集热图。C CRHBP CAP内皮细胞富集对患者预后的影响。D 来源于肿瘤和正常来源的细胞亚型之间的差异基因:左动脉内皮细胞;中毛细血管内皮细胞;右静脉内皮细胞。内皮细胞的 E UMAP 图,由 ENPP2 基因表达进行颜色编码。F 按 ENPP2 表达水平分层的患者预后。ISUP: International Society of Urological Pathology.UMAP: 均匀流形近似和投影+
ccRCC微环境中的空间代谢活性图谱
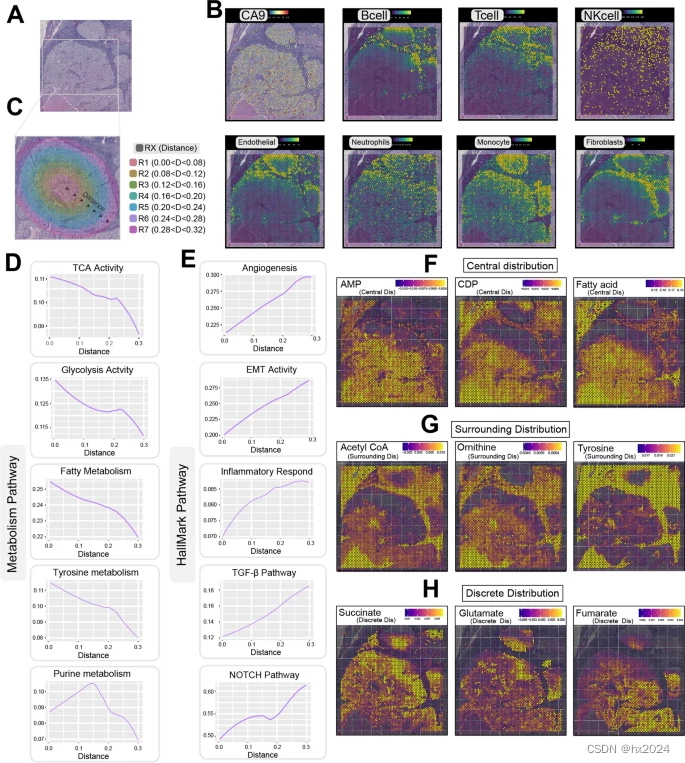 切片1:空间代谢活动的异质性。空间转录组学的病理学部分。B 各种细胞类型的近似分布。C 空间转录组学数据的分区,按与肿瘤中心的距离着色。D 代谢途径(糖酵解、TCA 循环、脂肪酸代谢、酪氨酸和嘌呤代谢)与距肿瘤中心距离的相关性。E 生物通路活性(EMT、血管生成、炎症、TGF-β 和 NOTCH)与距肿瘤中心距离的相关性。F 空间背景下 AMP、CDP 和脂肪酸平衡通量热图。G 空间背景下乙酰辅酶A、鸟氨酸和酪氨酸的平衡通量热图。H 空间背景下琥珀酸盐、谷氨酸盐和富马酸盐平衡通量热图。EMT—上皮-间充质转化
切片1:空间代谢活动的异质性。空间转录组学的病理学部分。B 各种细胞类型的近似分布。C 空间转录组学数据的分区,按与肿瘤中心的距离着色。D 代谢途径(糖酵解、TCA 循环、脂肪酸代谢、酪氨酸和嘌呤代谢)与距肿瘤中心距离的相关性。E 生物通路活性(EMT、血管生成、炎症、TGF-β 和 NOTCH)与距肿瘤中心距离的相关性。F 空间背景下 AMP、CDP 和脂肪酸平衡通量热图。G 空间背景下乙酰辅酶A、鸟氨酸和酪氨酸的平衡通量热图。H 空间背景下琥珀酸盐、谷氨酸盐和富马酸盐平衡通量热图。EMT—上皮-间充质转化
scMet算法
数据与代码
The single-cell RNA sequencing data by Chow J is available on the GEO database under Accession number GSE202374. Processed data from Saout JR’s single-cell RNA sequencing study can be accessed via the GEO portal with accession number GSE224630. Yu Z’s single-cell RNA sequencing data is accessible in the Gene Expression Omnibus datasets under accession number GSE207493. Li R’s single-cell RNA sequencing data can be downloaded as h5ad objects from Mendeley Data at Home PageMapping single cell transcriptomes in kidney cancer - Mendeley Data. Kim MC’s single-cell sequencing data has been deposited as GSE121638. Zhang Y’s single-cell RNA sequencing data is deposited in the National Center for Biotechnology Information Gene Expression Omnibus under Accession number GSE159115. Obradovic A’s single-cell sequencing data is available at GitHub - Aleksobrad/single-cell-rcc-pipeline: Data files and code for analysis of single-cell ccRCC data for the manuscript "Tumor-Specific Cell Populations in Clear Cell Renal Carcinoma Associated with Clinical Outcome Identified Using Single-Cell Protein Activity Inference." Includes code for VIPER protein activity inference pipeline. Krishna C's single-cell sequencing data can be found at https://www.ncbi.nlm.nih.gov/sra/PRJNA705464. Braun DA’s data can be accessed through the Single Cell Portal at Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma - Single Cell Portal. The spatial transcriptomics data of ccRCC is available on the Gene Expression Omnibus (GEO) database under the Accession number GSE175540. RNA-seq data for TCGA ccRCC and relevant clinical data are obtainable from The Cancer Genome Atlas Program (TCGA) - NCI. The immunohistochemistry images of clear cell renal cell carcinoma (ccRCC) can be found on https://www.proteinatlas.org/.The code for scFEA can be obtained from GitHub - changwn/scFEA: single cell Flux Estimation Analysis (scFEA) Try the below web server!. The code for scMet and R code for calculating cell infiltration enrichment can be obtained from GitHub - gwyang6/scMet: A package for deconvoluting Bulk RNAseq, using sc RNAseq to train CVAE models to generate new sc RNA seq data, and finally fitting Bulk RNAseq to obtain representative sc RANseq data















 240
240

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









