









数据来源
羊驼(好像是已经免疫过后的)外周血转录组/基因组经多重PCR扩增后,形成特定库并将这些序列重组于表达载体转入噬菌体(噬菌体展示技术),经固相/液相淘选后得到高亲和力的VHH序列库。该序列库再次放大构成高通量测序库,采用PE300测序策略。
实验目的
- paired reads 组装成productive contig
- 注释contig得到FWR1/CDR1/FWR2/CDR2/FWR3/CDR3/FWR4等信息
- 得到clonotype
- unique protein的统计信息,unqiue clonotype的统计信息等
MIXCR使用
安装mixcr
wget https://github.com/milaboratory/mixcr/releases/download/v3.0.13/mixcr-3.0.13.zip #https://github.com/milaboratory/mixcr/releases
unzip -d ~/software/mixcr mixcr-3.0.13.zip
echo "export PATH=~/software/mixcr/bin/mixcr:$PATH" > ~/.bashrc
source ~/.bashrc
overview
MIXCR的workflow主要包括三个步骤:
1、align:将reads与参考VDJC等基因对比
2、assemble:利用align的结果组装clonotype
3、export:将alignment或者clones的信息导出
4、input:fasta/fastq/fastq.gz/paired-end fastq/paired_end fastq.gz
5、output:mixcr的结果是二进制文件,需要使用exportAlignments and exportClones导出成易读格式
6、两种包装好的分析模式:analyze amplicon for analysis of targeted TCR/IG library amplification (5’RACE, Amplicon, Multiplex, etc). analyze shotgun for analysis of random fragments (RNA-Seq, Exome-Seq, etc).

示例
羊驼没有内置参考序列
MIXCR内置了鼠和人的reference
我的数据来源于alpaca,所以需要手动构建reference。#https://mixcr.readthedocs.io/en/latest/importSegments.html
#获取josn文件
wget https://github.com/repseqio/library-imgt/releases/download/v6/imgt.201946-3.sv6.json.gz
Library search path:
- built-in libraries
- /home/username/.
- /home/username/.mixcr/libraries
- /software/mixcr/libraries
所以我的josn文件丢在了/software/mixcr/libraries这里
#指定reference library
mixcr align --library imgt input_R1.fastq input_R2.fastq alignments.vdjca
#指定版本的reference library
mixcr align --library imgt.201631-4 input_R1.fastq input_R2.fastq alignments.vdjca
input
软件提供了一些用于控制input的参数
- –starting-material :初始建库材料 dna or rna
- –5-end :5‘端引物 no-v-primers or v-primers
- –3-end :3’端引物 j-primers or j-c-intron-primers or c-primers
- –adapters :有没有接头序列,有的话会帮我们做trim动作 ,adapters-present or no-adapters
output
.vdjca是align产生的二进制文件
.clns是asemble产生的二进制文件
# export alignments from .vdjca file
mixcr exportAlignments [options] alignments.vdjca alignments.txt
# export alignments from .clna file
mixcr exportAlignments [options] clonesAndAlignments.clna alignments.txt
# export clones from .clns file
mixcr exportClones [options] clones.clns clones.txt
# export clones from .clna file
mixcr exportClones [options] clonesAndAlignments.clna clones.txt
#customize the list of fields that will be exported by passing parameters to export commands
mixcr exportClones -count -vHit -jHit -vAlignment -jAlignment -aaFeature CDR3 clones.clns clones.txt
Analysis of targeted TCR/IG libraries
mixcr analyze amplicon -s alpaca \ #指定参考基因的种属,BCR只用人或鼠
--starting-material rna \ #建库最初时使用的扩增模板
--5-end v-primers --3-end j-primers \ #建库时的扩增引物
--adapters adapters-present \ #序列有没有测序用的adapter或者建库时的扩增引物
--library imgt
--receptor-type bcr \ #`tcr`, `bcr`, `tra`, `trb`, `trg`, `trd`, `igh`, `igk`, `igl`
--contig-assembly \ #要不要组装contig #store initial reads in the resulting `.vdjca` file
--only-productive
../NB244-R1-H_S1_L001_R1_001.fastq.gz ../NB244-R1-H_S1_L001_R2_001.fastq.gz \input
analysis1 #prefix of output
–starting-material affects the choice of V gene region which will be used as target in align step (vParameters.geneFeatureToAlign, see align documentation): rna corresponds to the VTranscriptWithout5UTRWithP and dna to VGeneWithP (see Gene features and anchor points for details).
#其实VGeneWithP == {UTR5Begin:VEnd} + {VEnd:VEnd(-20)}
VTranscriptWithout5UTRWithP == {L1Begin:L1End} + {L2Begin:VEnd} + {VEnd:VEnd(-20)}
#产生文件如下


High quality full length IG repertoire analysis
mixcr analyze amplicon \
--species hs \
--starting-material rna \
--5-end v-primers \
--3-end j-primers \
--adapters adapters-present \
--receptor-type BCR \
--region-of-interest VDJRegion \
--only-productive \
--align "-OreadsLayout=Collinear" \
--assemble "-OseparateByC=true" \
--assemble "-OqualityAggregationType=Average" \
--assemble "-OclusteringFilter.specificMutationProbability=1E-5" \
--assemble "-OmaxBadPointsPercent=0" \
input_R1.fastq input_R2.fastq analysis2
##############################################################################################################################
#cluster步骤,我们把searchdepth设置为0是不是VDJ序列完全一致的被聚到了一起
mixcr analyze amplicon \
--species hs \
--starting-material rna \
--5-end v-primers \
--3-end j-primers \
--adapters adapters-present \
--receptor-type BCR \
--region-of-interest VDJRegion \
--only-productive \
--align "-OreadsLayout=Collinear" \
--assemble "-OcloneClusteringParameters.searchDepth=0" \
--assemble "-OseparateByC=true" \
--assemble "-OqualityAggregationType=Average" \
--assemble "-OclusteringFilter.specificMutationProbability=1E-5" \
--assemble "-OmaxBadPointsPercent=0" \
input_R1.fastq input_R2.fastq analysis3
##############################################################################################################
问题
MIXCR的clonotype如何定义的?
读完说明书我认为是MIXCR的clonotype定义为CDR3 NDA序列完全一样的那些归为一个clonotype
它cluster之后的那些序列也不是我们通常意义上的clonotype(same of V and J reference gene and similarity of CDR3_aa >= 80% )
它的cluster也是根据DNA sequence进行的聚类
mixcr assemble [options] alignments.vdjca output.clns #在assemble过程中构建clonotype
mixcr assemble [options] -a alignments.vdjca output.clna # the outputs result in a “clones & alignments” format, allowing subsequent contig assembly
具体过程如下:

- 第一步:alignment结果文件中提取clonal sequence specified by assemblingFeatures parameter (CDR3 by default);如果aligned read 不包含clonal sequence 则会被丢弃
- 如果clonal sequence 包含低质量碱基,则会根据 badQualityThreshold 以及 maxBadPointsPercent 过滤
- After clonotypes are assembled by initial assembler and mapper, MiXCR proceeds to clustering。聚类时,clonotype with small counts will be attached to highly similar “parent” clonotypes with significantly greater count. After all clusters are built, only their heads are considered as final clones.

alignment是先组装还是先比对再组装?
- Before PE-read alignment:overlap > 17bp,minimal identity, minimal fraction of matching nucleotides between sequences >=0.9
- After PE-read alignment: 在不符合merge阈值时,但是两个reads比对到相同的V and J 基因时启动 Alignment-aided overlaps 进行reads的merge
alignment遇到低质量reads怎么办?
没有明说或者时我没有仔细看到,所以最好再运行mixcr时自己做质检以及过滤等动作
clonotype如何自定义?
One of the key MiXCR features is ability to assemble clonotypes by sequence of custom gene region (e.g. FR3+CDR3);
target clonal sequence can even be disjoint.
This region can be specified by assemblingFeatures parameter, as in the following example:
mixcr assemble -OassemblingFeatures="[V5UTR+L1+L2+FR1,FR3+CDR3]" alignments.vdjca output.clns
如下时assemble的控制参数:

Separation of clones with same CDR3 (clonal sequence) but different V/J/C genes

Clustering strategy:control clustering procedure are placed in cloneClusteringParameters parameters group which determines the rules for the frequency-based correction of PCR and sequencing errors:

如何理解Assemble full TCR/Ig receptor sequences
原文:MiXCR allows to assemble full TCR/Ig receptor sequences (that is all available off-CDR3 regions) with the use of assembleContigs command. Full sequence assembly may be performed after building of initial alignments and assembly of ordinary CDR3-based clonotypes.
个人理解:MIXCR 中assemble是assemble clones,是将相同clonal sequence的序列归为一个clonotype的动作,所以 full receptor assembly 应该是将整个抗体序列作为clonal sequence
https://mixcr.readthedocs.io/en/latest/assembleContigs.html
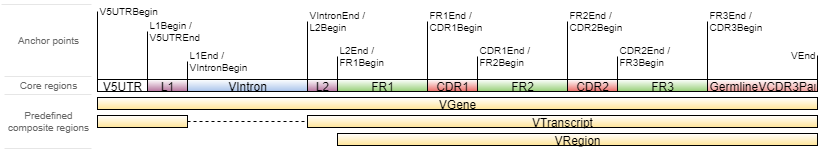
gene feature
The key feature of MiXCR is the possibility to specify:
- regions of reference V, D, J and C genes sequences that are used in alignment of raw reads
- regions of sequence to be exported by exportAlignments
- regions of sequence to use as clonal sequence in clone assembly
- regions of clonal sequences to be exported by exportClones
V Gene structure
D Gene structure

J Gene structure

















 452
452











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









