







本期教程
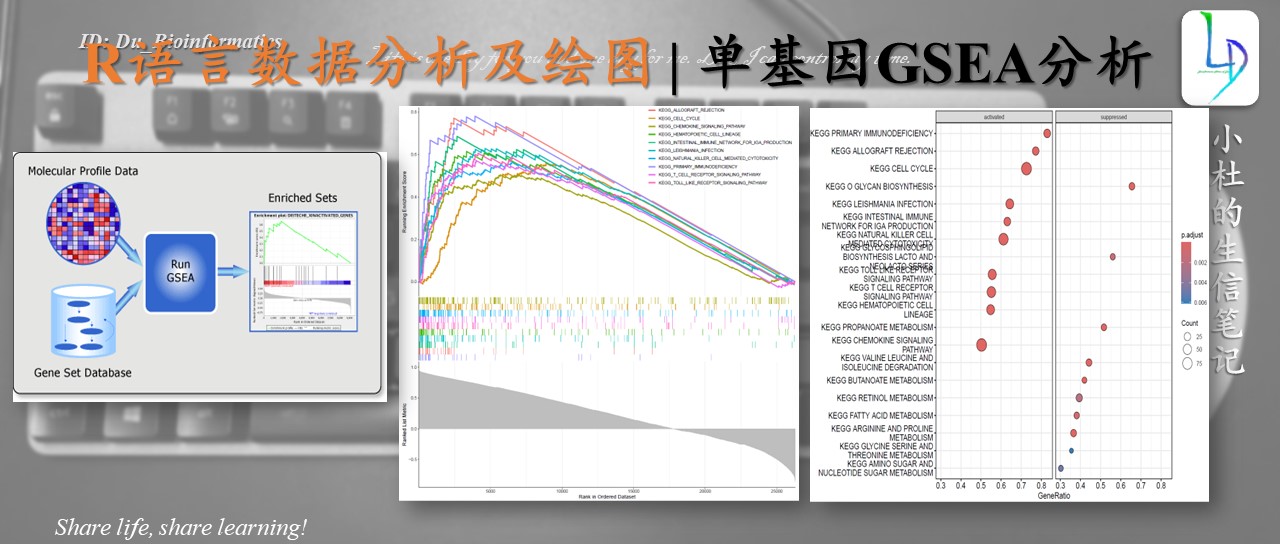
本期教程原文:一文掌握单基因GSEA富集分析 | gseaGO and gseaKEGG
写在前面
关于GSEA分析,我们在前期的教程单基因GSEA富集分析 | 20220404有出过类似的分享。今天,我们也结合相关的资源整理出一篇关于GSEA的教程及出图教程。每个方法的教程很多,我们大家结合自己的需求进行分析即可。以及,对于目前知识分享博主很多,只要你自己动手搜索,基本可以找到你的需求。
更新!…
对于GSEA的教程原计划是在2月2日发表,但是由于有预约被占用了,因此这个教程也就是往后推迟。我在2月1日将我们的教程发在社群中,**也有同学提出疑问:**单基因也可以做GSEA分析,以及给出自己的疑问?

也正是有了同学的疑问,才有今天后面更新的内容。也算进一步的了解GSEA分析原理以及单基因如何做GSEA?
对于社群,我认为这是一个值得鼓励的事情,针对自己的疑问提出疑问。以及在社群可以进行“激烈”讨论,这才是社群该有的“样子”,以及是我心中理想的社群该有的状态。
我们来这里看到这篇推文的很多同学,也许自己加了很多个社群,但是有用的社群也许也就那么几个而已(PS:包括自己)。小杜心中一直坚持的社群:不止是一个提供代码和数据的社群,而是一个可以“交流”的社群。社群的维护不在于建立社群的人,而是在于每一个人。
但是,又有多少社群可以一直保持这样的状态呢?有?但是很少。 但我们一直在路上…
OK!说的“废话太多”了。
对于GSEA的原理自己前面了解的也是比较局限。因此,自己也在进行学习,重新整理一下自己的知识结构。很多的知识总结,很多大佬都有总结。
若我们的分享对你有用,希望您可以点赞+收藏+转发,这是对小杜最大的支持。
GSEA原理
GSEA提出
GSEA(Gene set enrichment analysis)最先是2003年发表在Nature Genetics,题目为"PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes",DOI: 10.1038/ng1180。

此外,在2005年发表在Proc Natl Acad Sci USA,题目为:Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles,DOI: 10.1073/pnas.0506580102。

GSEA网址
https://www.gsea-msigdb.org/gsea/index.jsp

分析原理
GSEA的分析原理,我们这里使用“生信宝典”陈同老师分享的教程,一文掌握GSEA,超详细教程。
1. GSEA定义
GSEA用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),二是表达矩阵,软件会对基因根据其于表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
2. GSEA原理
给定一个排序的基因表L和一个预先定义的基因集S (比如编码某个代谢通路的产物的基因, 基因组上物理位置相近的基因,或同一GO注释下的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。

3. GSEA计算的关键概念
- 计算富集得分 (ES, enrichment score). ES反应基因集成员s在排序列表L的两端富集的程度。计算方式是,从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在s里面的基因,则增加统计值。遇到一个不在s里面的基因,则降低统计值。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度)是相关的。富集得分ES最后定义为最大的峰值。正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。

- 评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算p-value。

- 多重假设检验矫正。首先对每个基因子集s计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES)。随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值)

- Leading-edge subset,对富集得分贡献最大的基因成员。
4. 与GO或KEGG富集分析的区别
GO富集分析是先筛选差异基因,再判断差异基因在哪些注释的通路存在富集;这涉及到阈值的设定,存在一定主观性并且只能用于表达变化较大的基因,即我们定义的显著差异基因。
GSEA则不局限于差异基因,从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响。
以上是基于陈同老师的总结,对于GSEA概念与原理有初步的了解。
那么对于本教程的单基因GSEA分析如何做呢?
单基因GSEA分析
疑问
- 单基因GSEA是使用单个基因进行富集吗?
- 单基因GSEA的可靠性高吗?
…
也许我们大家有很多的疑问,同样自己也有很多的疑问。这就对了,我们在分析中,有疑问才有思考,有思考,可能对你的分析更有帮助。
在今天的教程中,我们一是给出(解答)大家的疑问;二是给大家对此有思考;三是,我们一起进行回答;四是,大家一起进行总结。
单基因GSEA原理
- 来源自己的愚见
单基因GSEA并不是说只是使用一个基因(目标基因)进行富集分析,而是通过目标基因与数据集基因进行关联分析,获得与目标基因高相关的数据集,进而使用此数据集进行富集分析。
对于单基因GSEA分析,主要针对的基因是未知功能的基因(PS:个人见解)。我们通过单基因GSEA分析,预测其目标基因的可能具有的功能,进而可以通过实验进一步验证。
- 如何做??
在我们前期的教程单基因GSEA富集分析主要基于表达量矩阵,计算目标基因与矩阵中其他基因的相关性,获得GSEA富集矩阵。本次的教程,也是使用这样的思路进行单基因GSEA分析。
GSEA数据集和软件下载
GSEA软件下载
提供不同平台的版本,但是我们这里依旧是推荐使用R语言进行分析,为什么呢?方便,代码直接有,粘贴复制即可。
https://www.gsea-msigdb.org/gsea/downloads.jsp
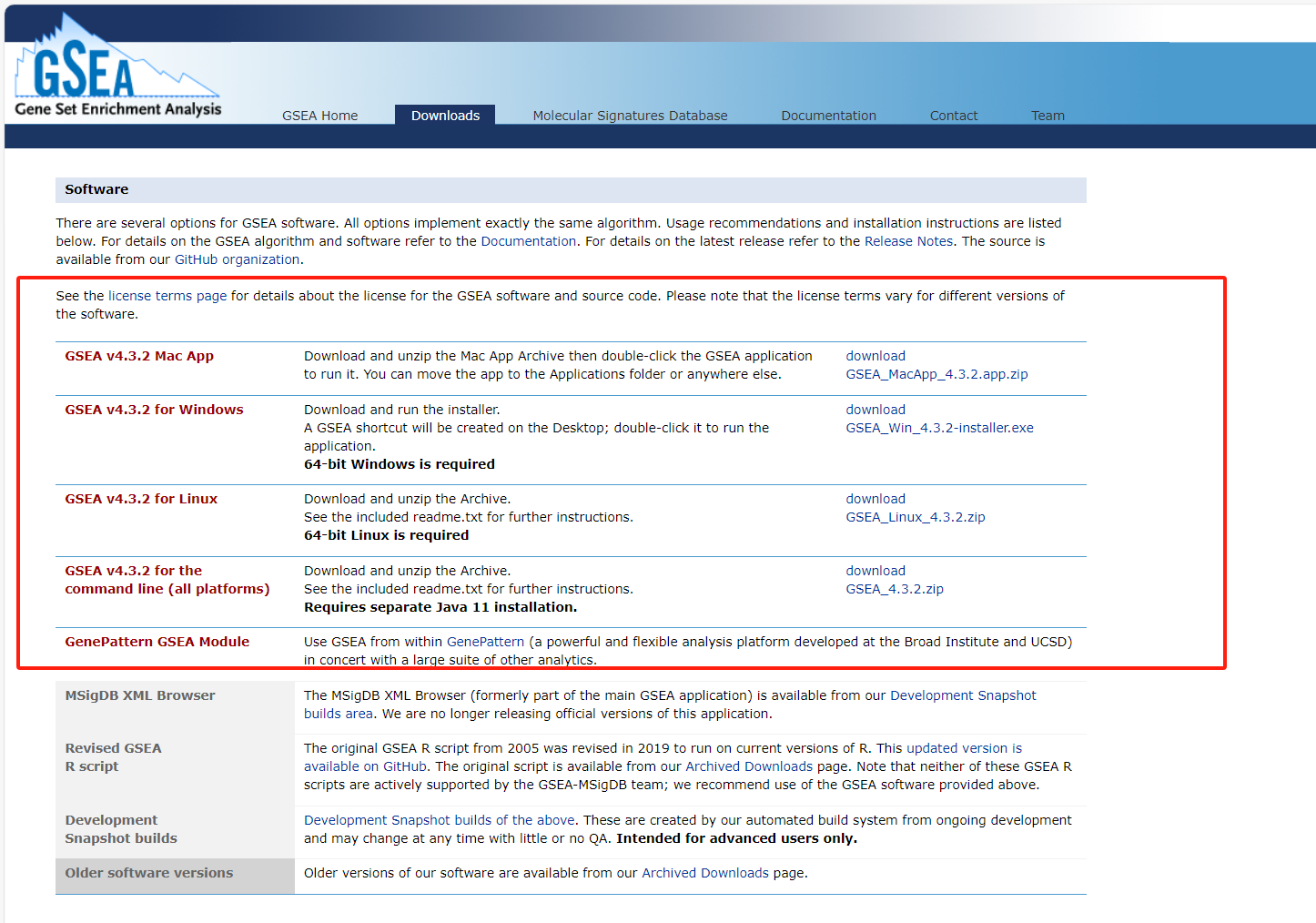
MsigDB数据集下载
网址:
https://www.gsea-msigdb.org/gsea/downloads.jsp
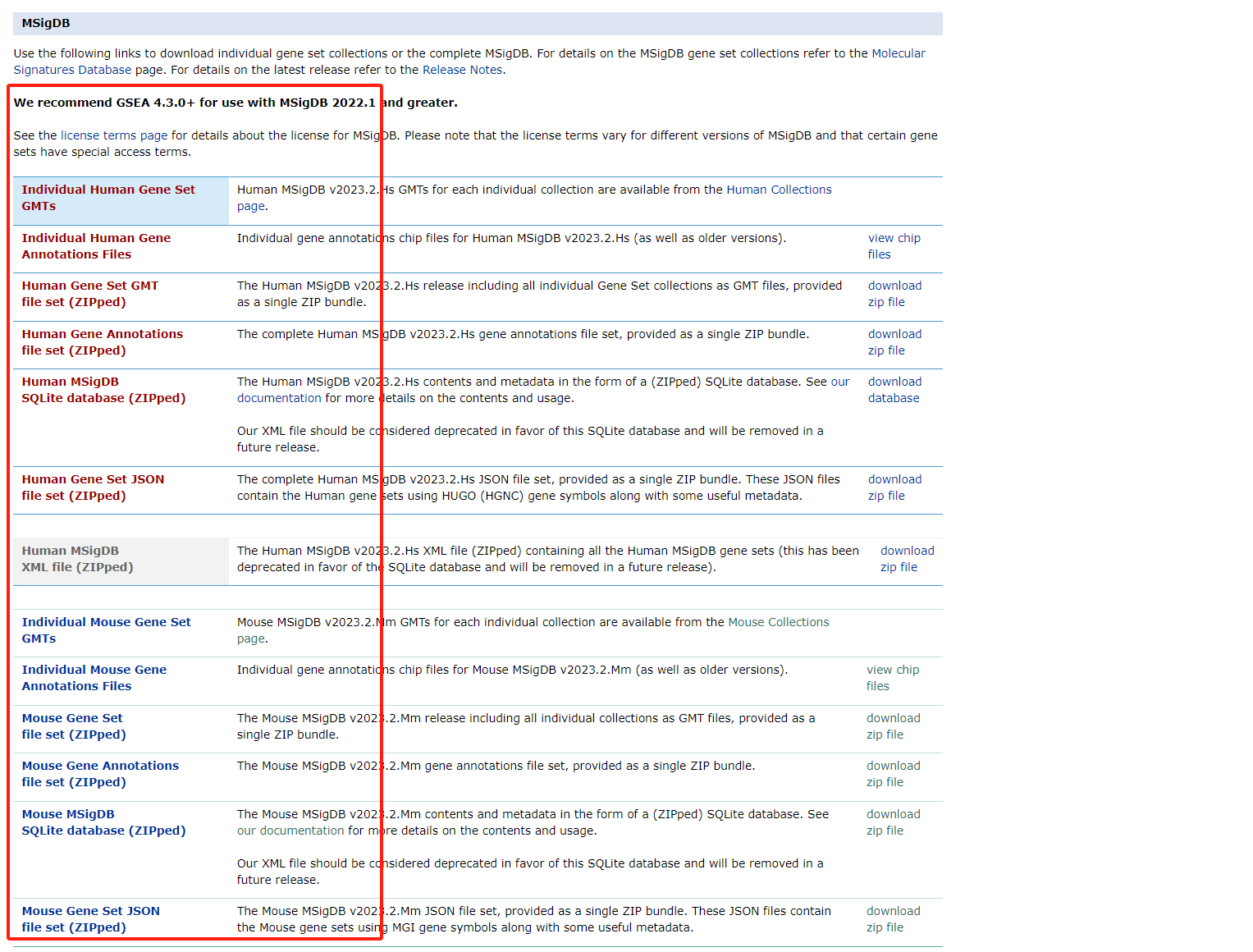
Human MSigDB Collections
https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp'
参考:
- https://www.gsea-msigdb.org/gsea/index.jsp
- https://mp.weixin.qq.com/s?__biz=MzI5MTcwNjA4NQ==&mid=2247484582&idx=1&sn=1e01276e1216c91bd6e1e08db15bd905&scene=21#wechat_redirect
- https://zhuanlan.zhihu.com/p/76160770
- https://zhuanlan.zhihu.com/p/625123340
单基因GSEA分析
输入数据类型
对于输入数据,基本是我们的基因数据集或是DEGs数据集。
绘图代码
导入所需的R包
###'@导入所需的R包
library(org.Hs.eg.db)
library(stringr)
library(BiocGenerics)
library(clusterProfiler)
library(enrichplot)
library(future)
library(future.apply)
导入数据
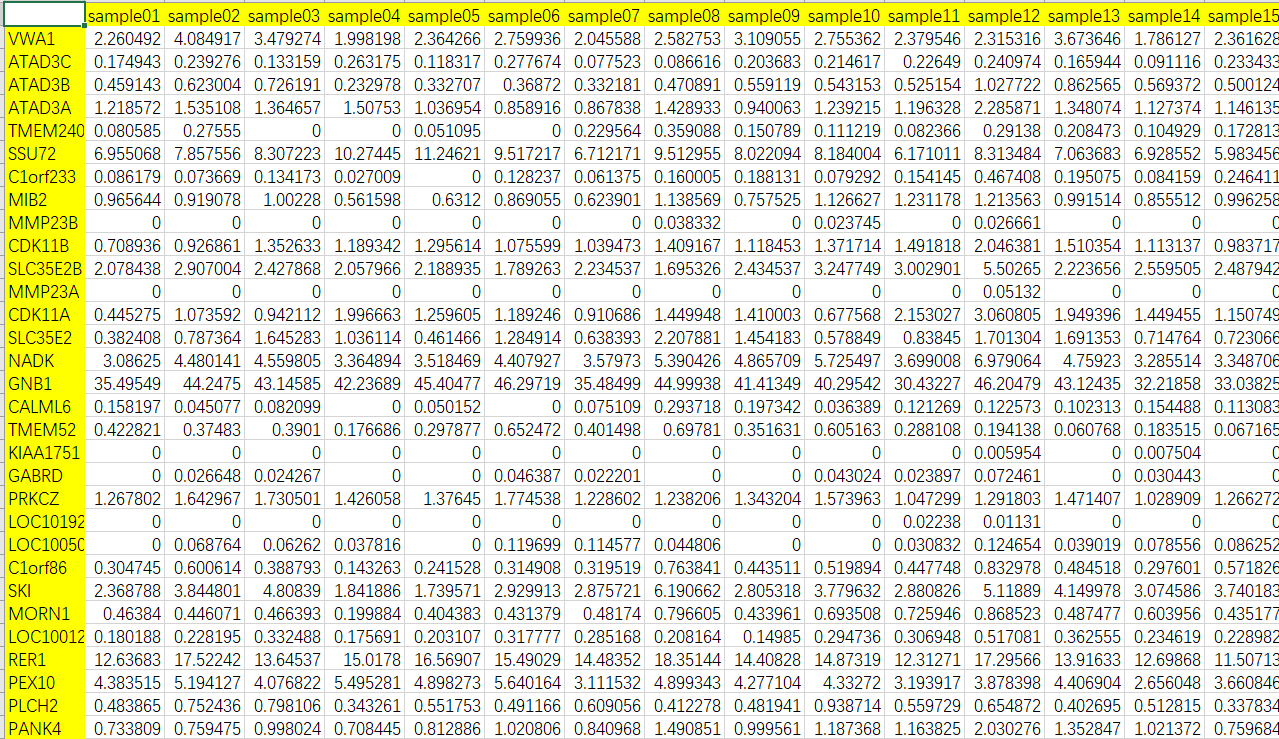
exprSet <- read.csv("Input_ExpData.csv",row.names = 1)
exprSet[1:10,1:5]
> exprSet[1:10,1:5]
sample01 sample02 sample03 sample04 sample05
VWA1 2.26049224 4.08491679 3.4792744 1.99819783 2.36426617
ATAD3C 0.17494291 0.23927633 0.1331588 0.26317521 0.11831695
ATAD3B 0.45914335 0.62300446 0.7261913 0.23297821 0.33270716
ATAD3A 1.21857160 1.53510784 1.3646565 1.50753047 1.03695410
TMEM240 0.08058546 0.27554979 0.0000000 0.00000000 0.05109502
SSU72 6.95506819 7.85755571 8.3072225 10.27444893 11.24621189
C1orf233 0.08617899 0.07366901 0.1341729 0.02700902 0.00000000
MIB2 0.96564412 0.91907833 1.0022802 0.56159762 0.63120040
MMP23B 0.00000000 0.00000000 0.0000000 0.00000000 0.00000000
CDK11B 0.70893552 0.92686092 1.3526327 1.18934155 1.29561449
计算相关性
GSEA分析简单逻辑就是:根据目标基因(单个基因)与其他基因之间的相关性,获得与目标基因具有高相关的基因集,再根据基因集进行GO和KEGG富集分析。(PS:这是个人的理解,若有错误,请根据自己的理解分析即可)
##'@计算相关性
##'@定义函数:获得目标基因与其他基因之间的P值和Cor值
batch_cor <- function(gene){
y <- as.numeric(exprSet[gene,])
rownames <- rownames(exprSet)
do.call(rbind,future_lapply(rownames, function(x){
dd <- cor.test(as.numeric(exprSet[x,]),y,type="spearman")
data.frame(gene=gene,mRNAs=x,cor=dd$estimate,p.value=dd$p.value )
}))
}
计算P值和Cor值
dd <- batch_cor("HES5")
提取基因集
gene <- dd$mRNAs
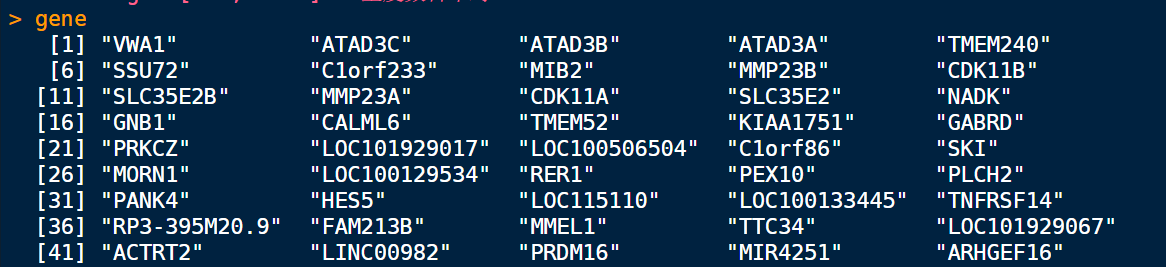
转化symbol格式
gene = bitr(gene, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
gene_df <- data.frame(cor=dd$cor,SYMBOL = dd$mRNAs)
去重复
gene <- dplyr::distinct(gene,SYMBOL,.keep_all=TRUE)
geneList <- gene_df$cor
names(geneList)=gene_df$SYMBOL
geneList=sort(geneList,decreasing = T)
dotplot_internal <- function(object, x = "GeneRatio", color = "pvalue",
showCategory=10, size=NULL, split = NULL,
font.size=12, title = "", orderBy="x", decreasing=TRUE) {
colorBy <- match.arg(color, c("pvalue", "p.adjust", "qvalue"))
if (x == "geneRatio" || x == "GeneRatio") {
x <- "GeneRatio"
if (is.null(size))
size <- "Count"
} else if (x == "count" || x == "Count") {
x <- "Count"
if (is.null(size))
size <- "GeneRatio"
} else if (is(x, "formula")) {
x <- as.character(x)[2]
if (is.null(size))
size <- "Count"
} else {
## message("invalid x, setting to 'GeneRatio' by default")
## x <- "GeneRatio"
## size <- "Count"
if (is.null(size))
size <- "Count"
}
df <- fortify(object, showCategory = showCategory, split=split)
## already parsed in fortify
## df$GeneRatio <- parse_ratio(df$GeneRatio)
if (orderBy != 'x' && !orderBy %in% colnames(df)) {
message('wrong orderBy parameter; set to default `orderBy = "x"`')
orderBy <- "x"
}
if (orderBy == "x") {
df <- dplyr::mutate(df, x = eval(parse(text=x)))
}
idx <- order(df[[orderBy]], decreasing = decreasing)
df$Description <- factor(df$Description, levels=rev(unique(df$Description[idx])))
ggplot(df, aes_string(x=x, y="Description", size=size, color=colorBy)) +
geom_point() +
scale_color_continuous(low="red", high="blue", name = color, guide=guide_colorbar(reverse=TRUE)) +
## scale_color_gradientn(name = color, colors=sig_palette, guide=guide_colorbar(reverse=TRUE)) +
ylab(NULL) + ggtitle(title) + DOSE::theme_dose(font.size) + scale_size(range=c(3, 8))
}
GSEA GO富集分析
##'@GSEA GO 富集
##'读取go.gmt文件
GOgmt<-read.gmt("c5.go.v7.2.symbols.gmt")
##'@富集分析
GO <-GSEA(geneList,TERM2GENE = GOgmt)
气泡图
dotplot_internal(GO)
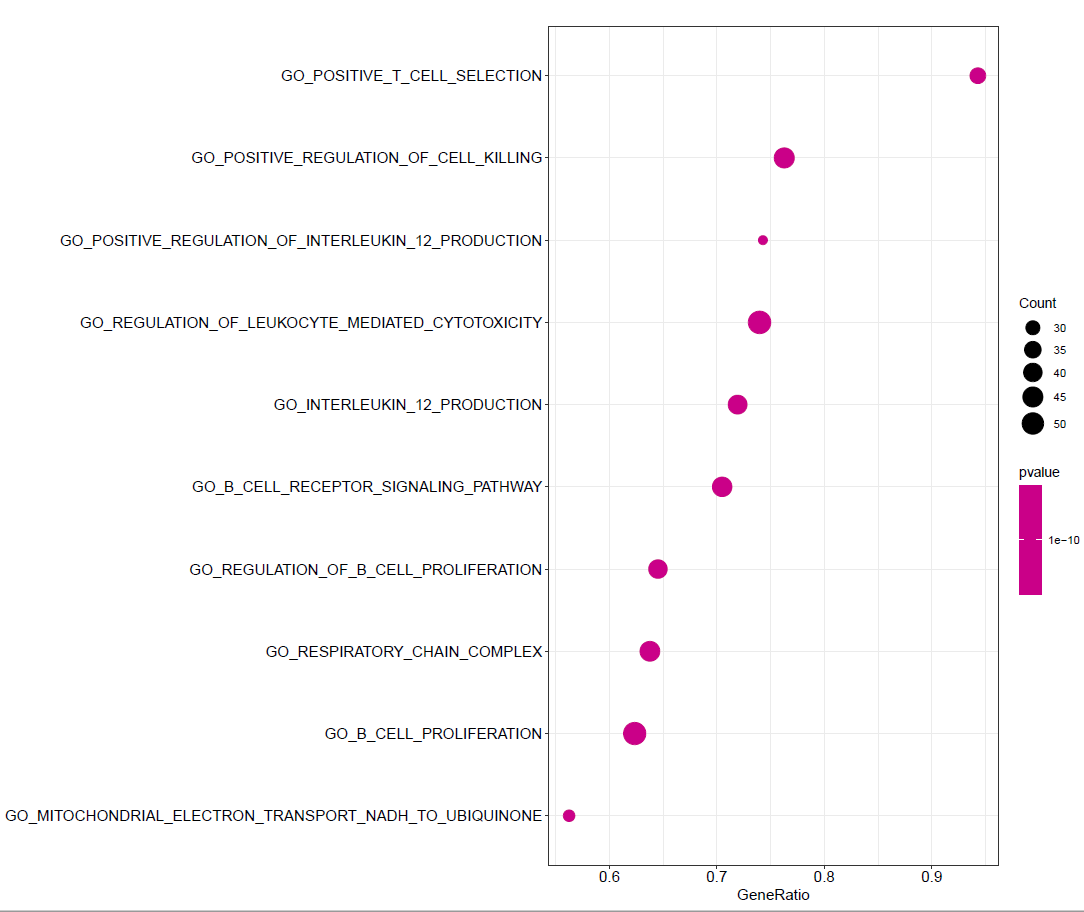
根据P值设置颜色
dotplot(GO,color="pvalue")
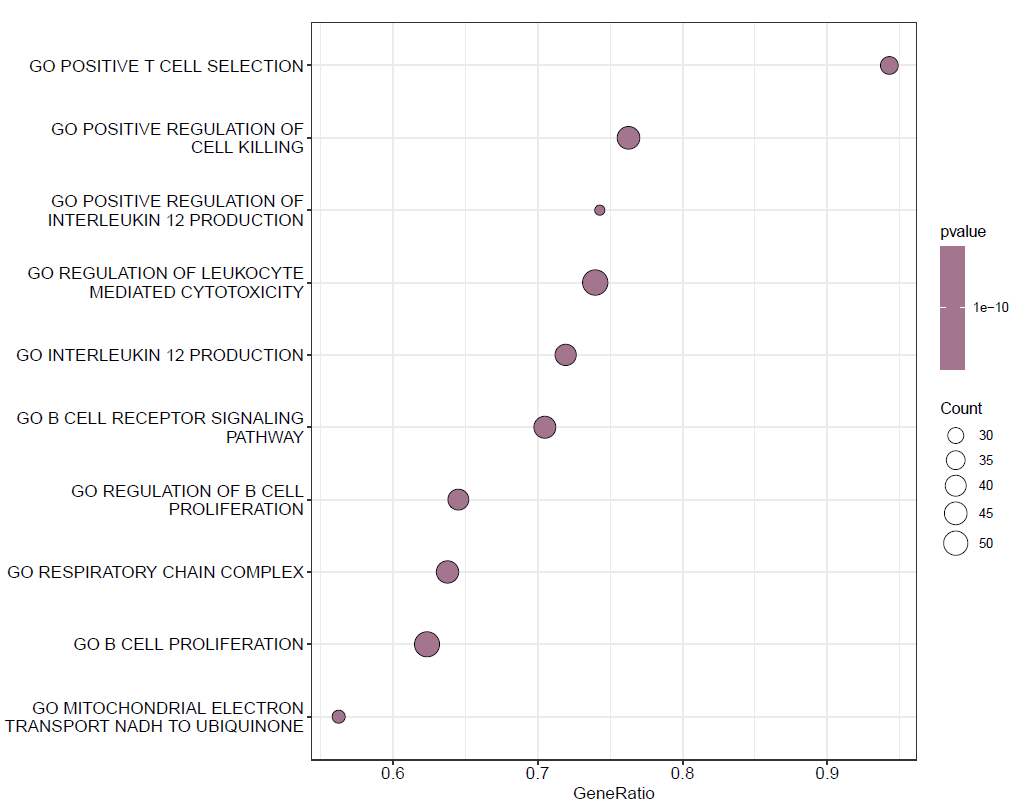
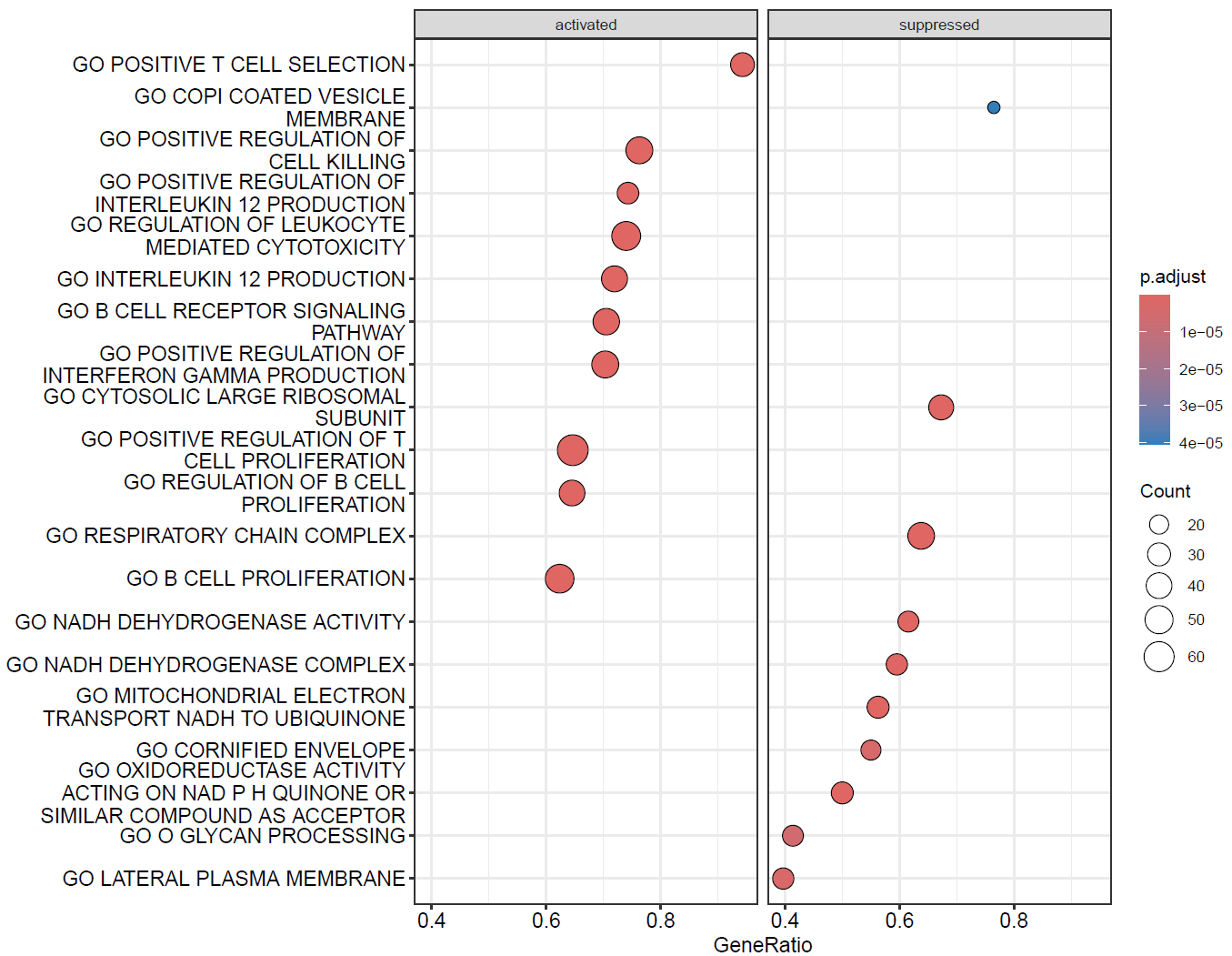
分类气泡图
dotplot(GO,split=".sign")+facet_grid(~.sign)
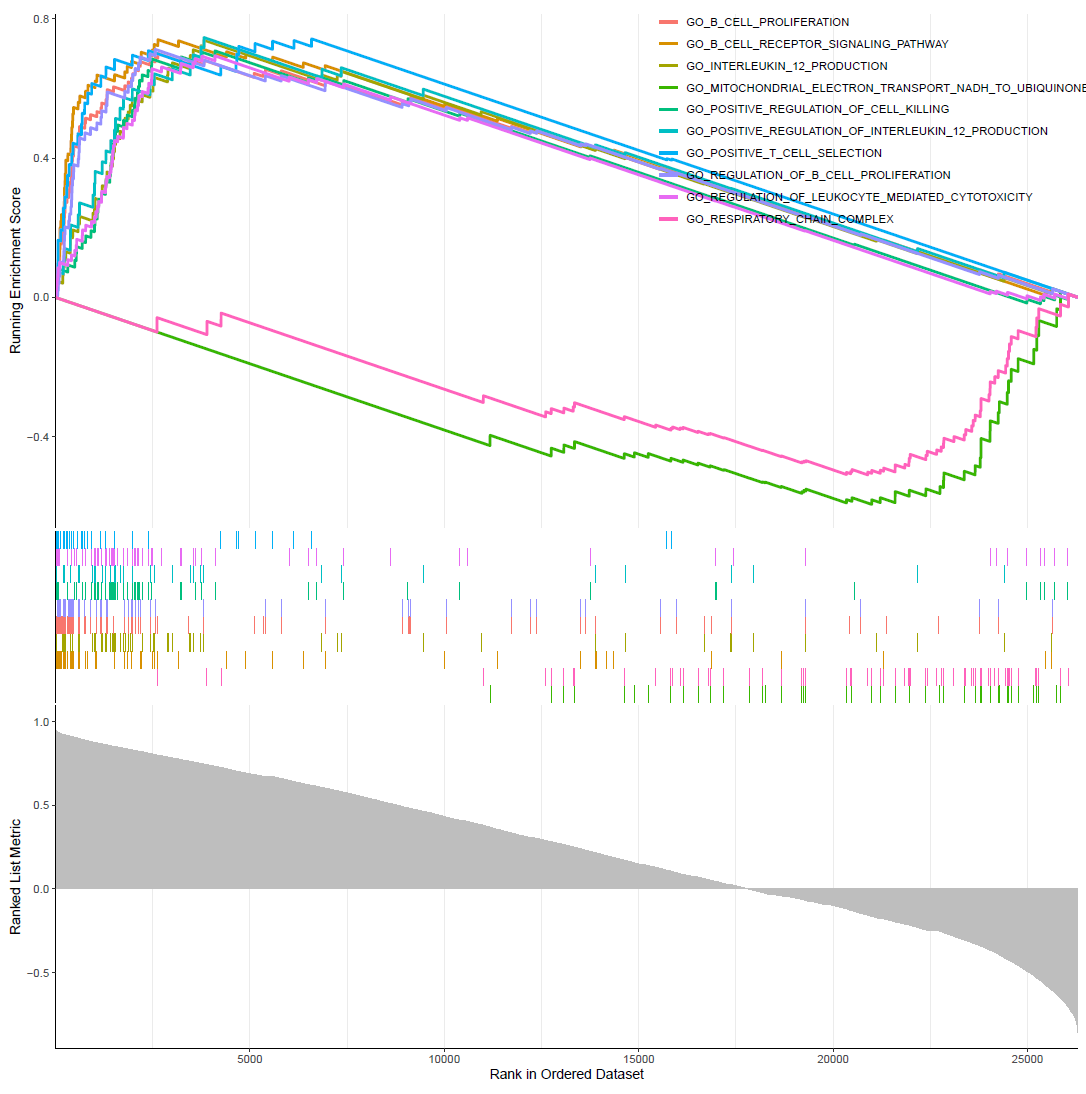
GSEA常规富集图
gseaplot2(GO,1:10,color="red",pvalue_table = F)
GSEA KEGG富集分析
导入kegg.gmt文件
KEGGgmt<-read.gmt("c2.cp.kegg.v7.2.symbols.gmt")
KEGG富集分析
KEGG <-GSEA(geneList,TERM2GENE = KEGGgmt)
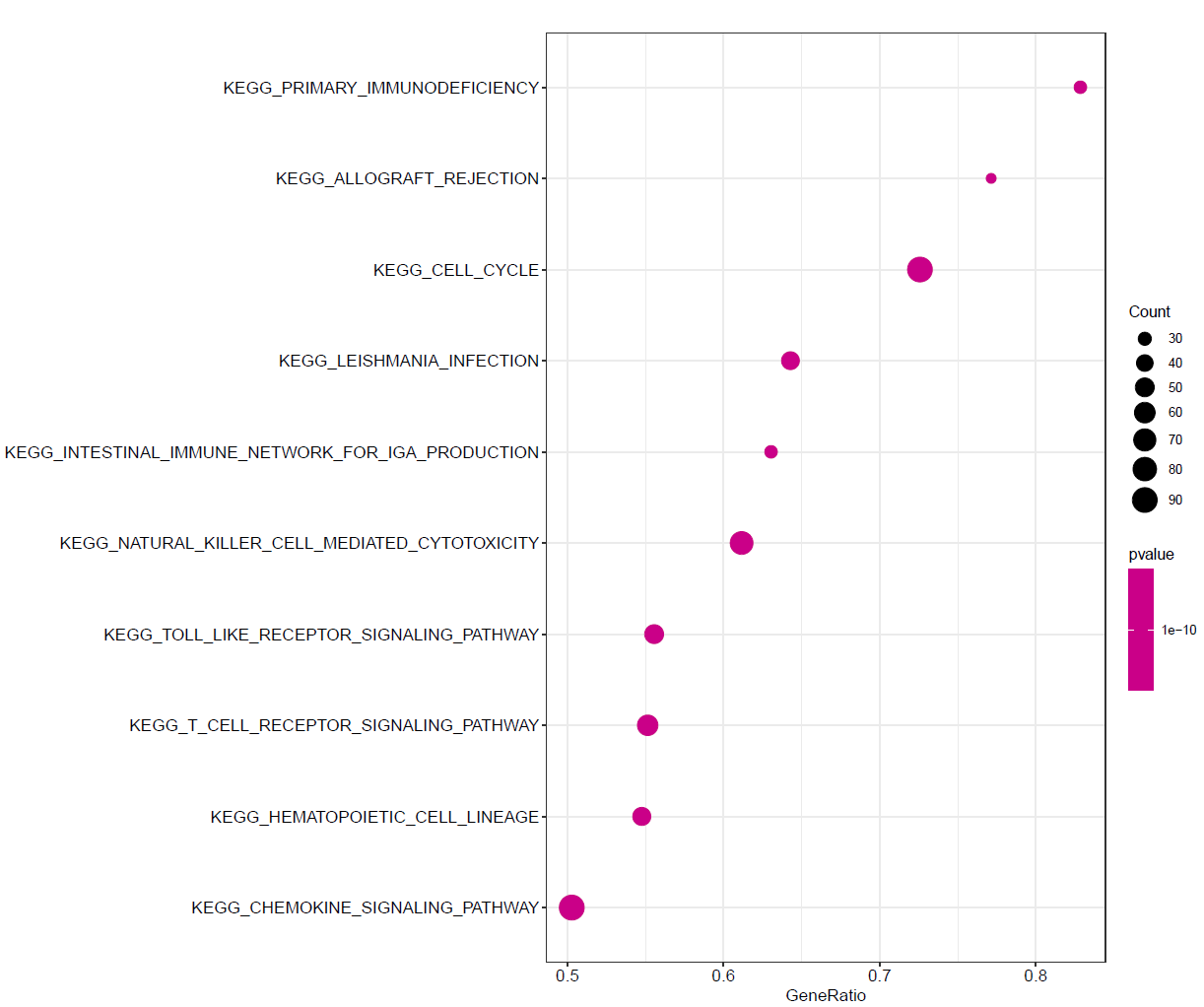
GSEA_KEGG气泡图
dotplot_internal(KEGG)
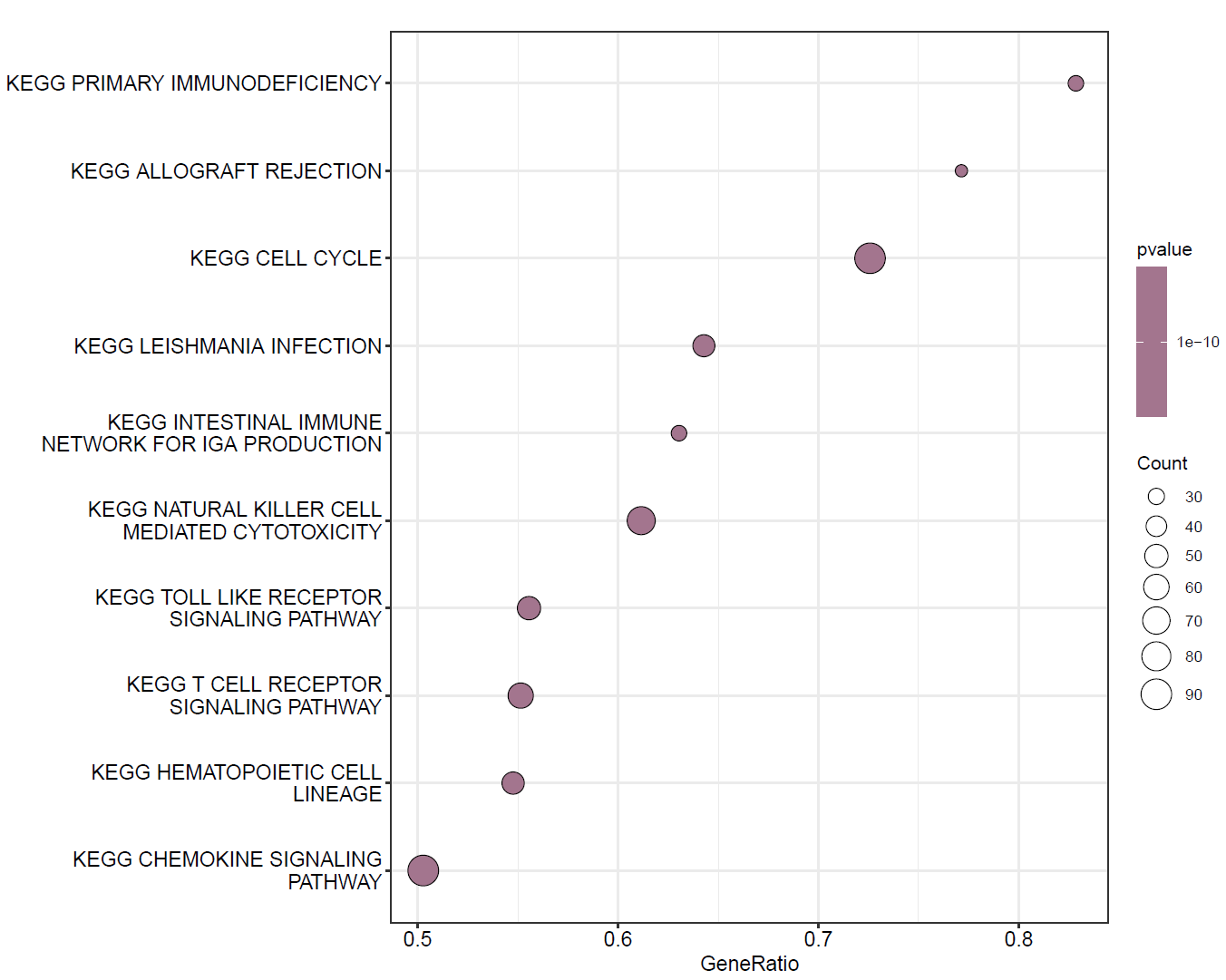
KEGG 气泡图(根据P值设置颜色)
dotplot(KEGG,color="pvalue")
KEGG 分类气泡图
dotplot(KEGG,split=".sign")+facet_grid(~.sign)

常规GSEA KEGG富集图
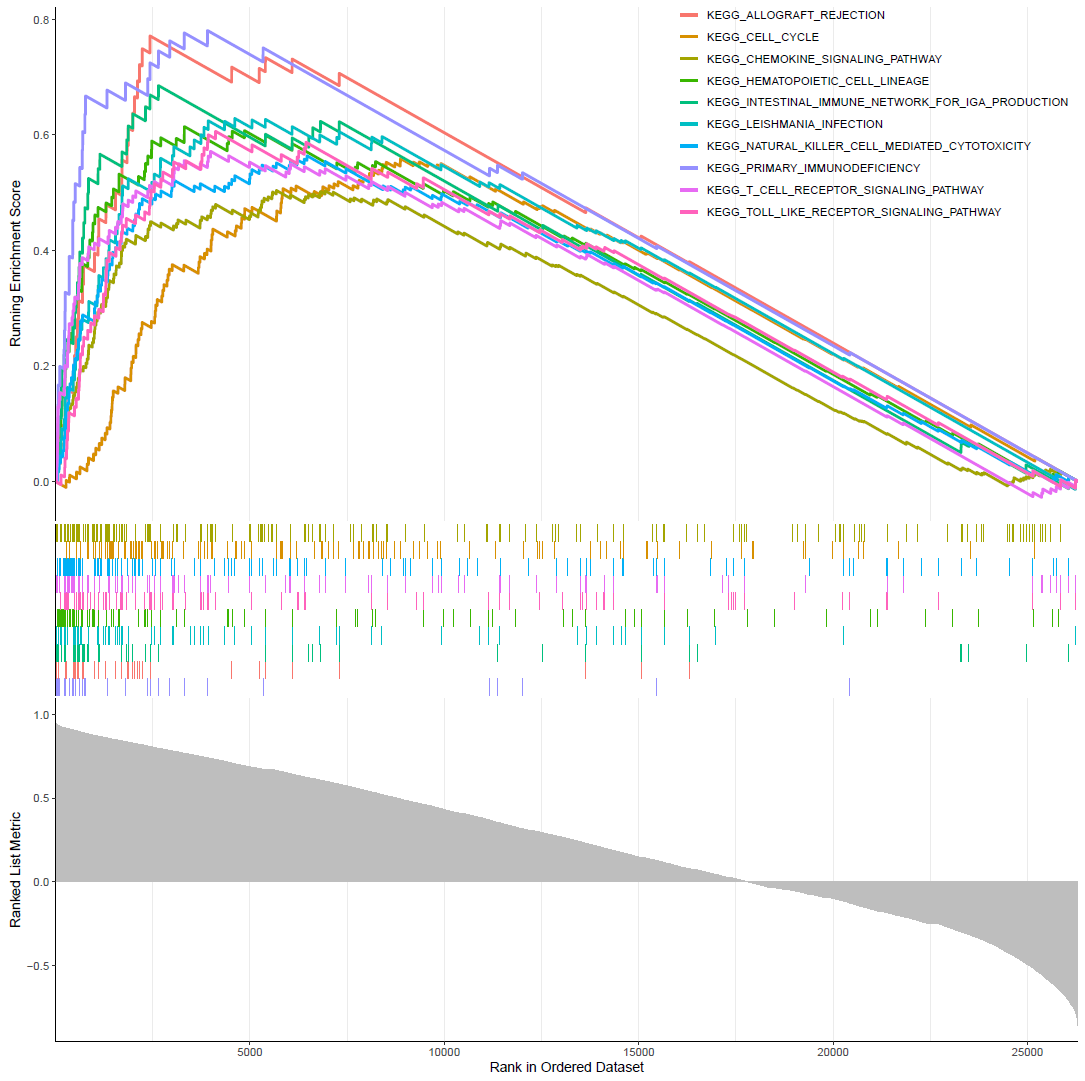
gseaplot2(KEGG,1:10,color="red",pvalue_table = F)
若我们的分享对你有用,希望您可以点赞+收藏+转发,这是对小杜最大的支持。
本期教程原文:一文掌握单基因GSEA富集分析 | gseaGO and gseaKEGG
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
4. 精美图形绘制教程
5. 转录组分析教程
一个转录组上游分析流程 | Hisat2-Stringtie
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!


















 8354
8354

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











