






在已有分子或者离子的lammps格式的拓扑文件的情况下,可以使用intermol快速生成gromacs格式的拓扑文件。intermol是一个python包,可以对多种分子动力学模拟软件使用的拓扑文件进行相互转化,请访问官网获取更多信息。本文以LiTFSI/PEO电解质体系为例,演示如何在linux下用intermol将lammps拓扑文件转化为可以使用的gromacs拓扑文件。
一、intermol的安装及使用
参考官方安装教程
1、检查环境
官方要求python版本>=2.7,但是版本太低有可能出现报错,这里建议python版本>=3.8,我这里采用3.8.5的版本。python环境中需要有numpy和parmed这两个包,如果使用了conda建立的虚拟环境,可以使用如下代码检查和安装numpy,parmed类似。
conda list|grep numpy #检查当前python环境是否安装numpy
conda install numpy #在当前环境安装numpy
2、安装intermol
在linux中适当路径下运行下面的代码
git clone https://github.com/shirtsgroup/InterMol.git #下载
cd InterMol
pip install . #安装
3、运行示例
cp intermol/convert.py ~/bin/
#将转化拓扑文件的脚本复制到~/bin/,方便寻找
cd intermol/tests/lammps/unit_tests/angle_permute-1_vacuum/
#进入测试文件夹
python ~/bin/convert.py --lmp_in angle_permute-1-data_vacuum.input --gromacs
#执行脚本开始转化
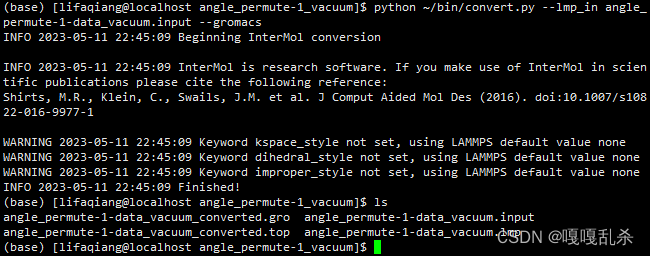
成功执行后会出现上面的输出,此时会多出后缀分别为gro和top的两个文件,分别是gromacs的坐标文件和拓扑文件,后缀为input和lmp的文件分别是lammps的in文件和data文件,均为这个脚本的输入文件。输入文件可以是其它的后缀甚至无后缀。准备输入文件时,可以参考这里的input和lmp文件的内容。
4、软件实操
我使用的in文件tfsi.input内容如下,参考了前面示例中的input文件,intermol要求in文件中用read_data引入data文件,我这里是tfsi.data,data文件中必须包含力场参数,并且与in文件中的力场设置一致。data文件的第一行要求是分子名,因为convert.py会把data文件第一行的内容作为.top文件的分子名。
units real
atom_style full
dimension 3
boundary p p p
pair_style lj/cut/coul/long 11 11
pair_modify mix geometric
bond_style harmonic
angle_style harmonic
special_bonds lj/coul 0 0 0.5
dihedral_style opls
read_data tfsi.data
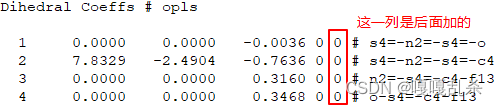
intelmol要求data文件中的Dihedral Coeffs的每一行要用5个系数来描述势函数,否则会报错IndexError: list index out of range。如果Dihedral Coeffs只用了4个系数可以在后面补0,如下图所示。
进入文件所在的目录输入下面的命令运行转化脚本生成tfsi_converted.gro和tfsi_converted.top
python ~/bin/convert.py --lmp_in tfsi.input --gromacs
把tfsi_converted.top重命名为tfsi.top,类似的方法可以获得PEO的拓扑文件peo.top和锂离子的拓扑文件li.top
二、输出文件解析
下面查看一下tfsi_converted.gro和tfsi_converted.top的内容
1、gro文件
Untitled ;标题
15 ;原子数
1R01 A1 1 -0.572679896300 0.129348522000 -0.115282596500
1R01 A2 2 -0.420477218700 0.102714013200 -0.004757462200
1R01 A3 3 -0.719493372200 0.182345141800 -0.006963637600
1R01 A4 4 -0.675471572700 0.342027425400 0.086131039000
1R01 A5 5 -0.375935995600 0.265865698700 0.081814181000
1R01 A6 6 -0.458996747800 -0.030030267100 0.125594774500
1R01 A7 7 -0.758794537400 0.044678755500 0.117971278400
1R01 A8 8 -0.873632374300 0.213941664800 -0.118622045000
1R01 A9 9 -0.271753917500 0.044740905400 -0.113150626400
1R01 Aa 10 -0.241579499100 0.148460353300 -0.215827724500
1R01 Ab 11 -0.306624502800 -0.083422857800 -0.180704712400
1R01 Ac 12 -0.152210019700 0.024312318000 -0.026551904900
1R01 Ad 13 -0.842768478500 0.322181835100 -0.216318927000
1R01 Ae 14 -0.908263207000 0.089042028000 -0.192205381800
1R01 Af 15 -0.988938589700 0.255110880600 -0.033647237300
0.9367286 0.5254503 0.4419137 ;盒子x,y,z方向的长度,单位为nm
gro文件为gromacs的原子坐标文件,与pdb文件不同的是gro文件不包含原子连接信息。中间的列从左到右依次为残基序号(1)、残基名称(R01)、原子名称、原子序号、原子坐标三列,有时候最后面会多出三列,分别是原子x,y,z方向的速度,单位为nm/ps。下面是top文件的段。
2、defaults段
[ defaults ] ;力场设置
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 3 yes 0.500000 0.500000
1、nbfunc为非键函数类型, 对应in文件的pair_style,取1(Lennard-Jones)或2(Buckingham)
2、comb-rule为组合规则,对应in文件的pair_modify,组合规则3对应in文件的mix geometric
3、gen-pairs确定是否自动生成1-4相互作用,默认为no。程序会优先读取[ pairs ]中的1-4相互作用,当[ pairs ]中没有1-4相互作用时,gen-pairs为yes就会自动生成1-4相互作用,gen-pairs为no就会报错
4、fudgeLJ为LJ势1–4相互作用的修正因子,默认为1,仅当gen-pairs被设置为‘yes’时才会使用fudgeLJ
5、fudgeQQ为静电1–4相互作用的修正因子,默认为1。special_bonds lj/coul 0 0 0.5指定LJ势和库伦势的1-4相互作用修正因子均为0.5,对应fudgeLJ和fudgeQQ
3、atomtypes段
[ atomtypes ] ;定义原子类型
;type, bondingtype, atomic_number, mass, charge, ptype, sigma, epsilon
lmp_001 lmp_001 -1 14.00700000 0.00000000 A 3.25000000e-01 7.10861600e-01
lmp_002 lmp_002 -1 32.06000000 0.00000000 A 3.55000000e-01 1.04600000e+00
lmp_003 lmp_003 -1 15.99900000 0.00000000 A 3.15000000e-01 8.37218400e-01
lmp_004 lmp_004 -1 12.01100000 0.00000000 A 3.50000000e-01 2.76144000e-01
lmp_005 lmp_005 -1 18.99800000 0.00000000 A 3.11800000e-01 2.55224000e-01
一般来说,[ atomtypes ]段会在gromacs自带的力场文件里,由拓扑文件include进去,由于这里的原子采用自定义的参数,所以需要加上这个段。
1、type为原子类型,下面的原子类型是程序自定义的
2、bondingtype为成键类型,下面的成键类型是程序自定义的
3、atomic_number为原子序数,lammps的拓扑文件不包含原子序数信息,所以程序无法得出原子序数,只能用-1表示,此参数对计算无影响
4、mass为相对原子质量,charge为原子电荷
6、ptype为粒子类型,A表示原子
7、sigma和epsilon为LJ势参数,单位分别为nm和kJ/mol
4、moleculetype段
[ moleculetype ] ;定义分子类型
tfsi 3
1、tfsi为分子名
2、3表示排除相邻的3个键的非键相互作用,这意味着排除1-2、1-3、1-4相互作用,但是其优先级低于[ defaults ]中的gen-pairs,所以1-4相互作用并没有被排除
5、atoms段
[ atoms ] ;定义原子
;num, type, resnum, resname, atomname, cgnr, q, m
1 lmp_001 1 R01 A1 0 -0.46200000 14.00700000
2 lmp_002 1 R01 A2 0 0.71400000 32.06000000
;这里省略下面的行,后面省略不再注释
1、num为原子编号
2、type为原子所属的原子类型,下面的原子类型已在atomtypes字段中定义
3、resnum为残基编号
4、resname为残基名称
5、atomname为原子名称
6、cgnr为电荷组编号,gromacs必须保证原子组的总电荷为0,这些组被称为电荷组
7、q为电荷,m为相对原子质量
6、pairs段
[ pairs ]
; ai aj funct
1 10 1
1 11 1
分子内原子对之间额外的LJ和静电相互作用可以添加到分子定义部分的[ pairs ]字段中,这些相互作用的参数可以独立于[ pairtypes ]中定义的非键相互作用参数进行设置。
1、ai、aj是pairs势两原子的编号,后文的ak、i、j、k、l类似
2、funct是函数形式,1对应普通对相互作用和1–4相互作用的函数类型
7、bonds段
[ bonds ]
; ai aj funct r k
1 2 1 1.57000000e-01 1.56849792e+05
1 3 1 1.57000000e-01 1.56849792e+05
funct=1即最常用的简谐势,r、k单位分别为nm、kJ mol − 1 ^{-1} −1 nm − 2 ^{-2} −2
8、angles段
[ angles ]
; ai aj ak funct theta cth
1 2 5 1 1.13600000e+02 7.89018720e+02
1 2 6 1 1.13600000e+02 7.89018720e+02
funct=1同样为简谐势,theta、cth单位分别为deg、kJ mol − 1 ^{-1} −1 rad − 2 ^{-2} −2
9、dihedrals段
[ dihedrals ]
; i j k l func
1 2 9 10 3 6.61072000e-01 1.98321600e+00 0.00000000e+00 -2.64428800e+00 -0.00000000e+00 -0.00000000e+00
1 2 9 11 3 6.61072000e-01 1.98321600e+00 0.00000000e+00 -2.64428800e+00 -0.00000000e+00 -0.00000000e+00
funct=3为Ryckaert-Bellemans二面角,程序自动将OPLS参数变换为Ryckaert-Bellemans参数C 0 _0 0、C 1 _1 1、C 2 _2 2、C 3 _3 3、C 4 _4 4、C 5 _5 5,其单位是kJ mol − 1 ^{-1} −1。
10、system段和molecules段
[ system ] #定义系统名
Untitled
[ molecules ] #定义系统中包含的分子
; Compound nmols
tfsi 1
molecules段的每个名称必须对应于拓扑文件前面的[ moleculetype ]中给出的名称,分子的顺序必须与坐标文件中的顺序完全一致,nmols定义包含的分子数量。
参考文献
https://jerkwin.github.io/GMX/GMXman-4/
https://jerkwin.github.io/GMX/GMXman-5/
三、修改输出文件
由于peo.top、tfsi.top、li.top三个文件中的原子类型和成键类型有重复,因此需要修改其中两个文件的原子类型和成键类型使其不重复,可以使用Notepad++的替换功能进行修改。
defaults段和atomtypes段属于力场定义,system段和molecules段属于体系定义,其余段是分子定义。为了使top文件变得简洁,分子定义部分会单独放在一个后缀为itp的文件里,由主top文件include进去。这里把peo.top、tfsi.top、li.top重命名为peo.itp、tfsi.itp、li.itp,再把defaults段、atomtypes段、system段、molecules段提取出来放到主top文件mix.top里,其内容如下。
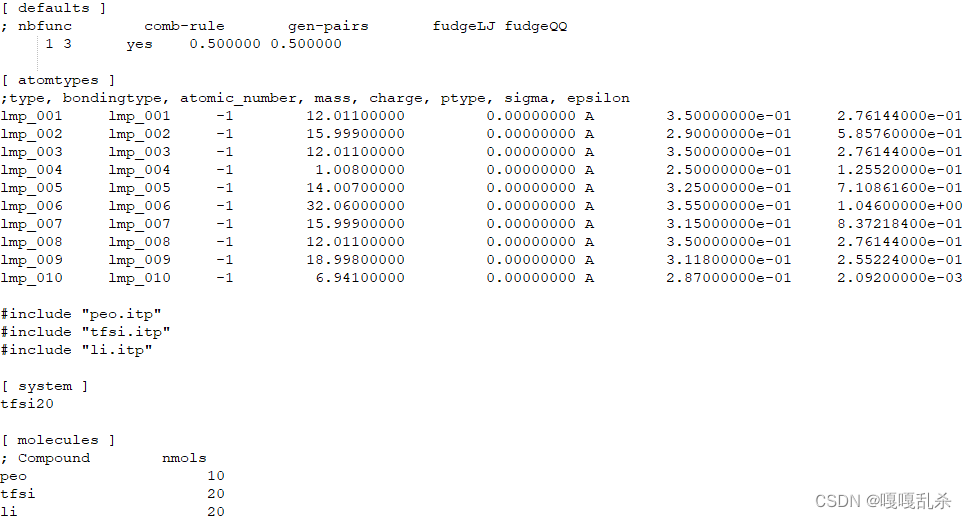
top文件段的顺序有严格的要求,可以参考这个帖子。另外,拓扑文件里的的原子顺序要和gro文件一致。我用的gro文件是packmol生成的pdb文件转化过来的,所以具体来说就是,molecules段里的分子顺序和数量要和inp文件一致。至此,gromacs的拓扑文件就制作好了。















 1638
1638

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









