







 该文详细介绍了基于TCGA数据的胃癌基因表达分析流程,包括数据预处理、火山图绘制、功能富集分析及通路富集。通过比较癌症和正常样本的基因表达差异,识别上调和下调基因,并探讨其在样本中的分布及表达差异。文章使用R语言进行分析,展示了基因在癌症发展中的潜在作用。
该文详细介绍了基于TCGA数据的胃癌基因表达分析流程,包括数据预处理、火山图绘制、功能富集分析及通路富集。通过比较癌症和正常样本的基因表达差异,识别上调和下调基因,并探讨其在样本中的分布及表达差异。文章使用R语言进行分析,展示了基因在癌症发展中的潜在作用。
一、数据的准备(本文以TCGA中胃癌数据为例)
#载入表达数据与注释数据
exp2 <- read.delim("f:/TCGA-STAD.htseq_fpkm.tsv")
anno2 <- read.delim("f:/gencode.v22.annotation.gene.probeMap")
#数据大小
> dim(exp2)
[1] 60483 408
> dim(anno2)
[1] 60483 6
#将ensembl名转换成gene id
geneID2 <- anno2[match(exp2$Ensembl_ID,anno2[,1]),1:2]
geneData2 <- cbind(geneID2$gene,exp2[,-1])
#重复探针表达值取均值
mean_data2 <- aggregate(geneData2[,2:dim(geneData2)[2]],by=list(gene=geneData2[,1]),FUN = mean)
#将基因名赋予行名
rownames(mean_data2) <- mean_data2[,1]
mean_data2 <- mean_data2[,-1]
> dim(mean_data2)
[1] 58387 407
> mean_data2[1:3,1:3]
TCGA.D7.5577.01A TCGA.D7.6818.01A TCGA.BR.4280.01A
5_8S_rRNA 0.17582972 0.7885477 0.00000000
5S_rRNA 0.04944558 0.1269647 0.06802885
7SK 0.00000000 0.0760312 0.02682956
#区分癌症样本和正常样本
STADtumor <- mean_data2[,which(as.numeric(substr(colnames(mean_data2),14,15))<=9)]
STADnormal <- mean_data2[,-which(as.numeric(substr(colnames(mean_data2),14,15))<=9)]
STADdata <- cbind(STADtumor,STADnormal)
> dim(STADtumor)[2]#癌症数目
[1] 375
> dim(STADnormal)[2]#正常数目
[1] 32
#数据预处理:保留至少在75%的样本中都有表达的基因
#数据已经FPKM处理,无需标准化
keepData <- rowSums(STADdata>0) >= floor(0.75*ncol(STADdata))
STADdata <- STADdata[keepData,]
> dim(STADdata)#剩余样本数
[1] 31047 407
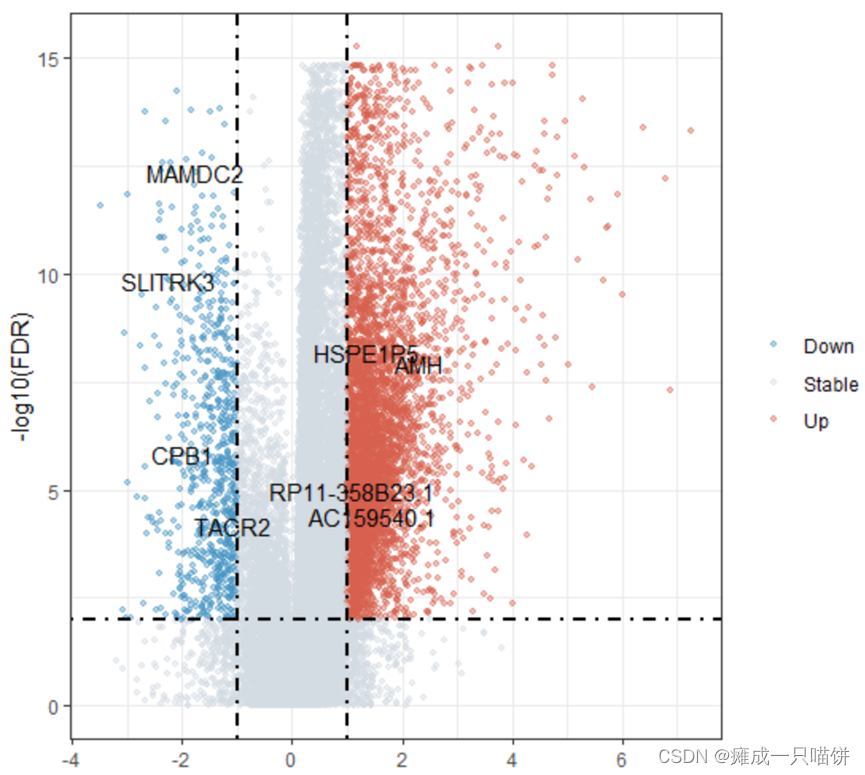
二、绘制火山图
所需数据准备:
#fc值计算
STADfc <- apply(STADdata, 1, function(x){mean(x[1:375]/mean(x[376:407]))})
#p值计算
STADpval <- apply(STADdata, 1,function(x) {wilcox.test(x[1:375],x[376:407])$p.value})
#p值校正
STADfdr <-p.adjust(STADpval,method = "BH")
#数据整理
STADvol <- data.frame(log2(STADfc),-log(STADfdr,10))
#卡阈值:fdr<0.01,abs(log(fc))>=1
STADvol[,3] <- ifelse(STADvol[,2] > 2 & abs(STADvol[,1]) >= 1,
ifelse(STADvol[,1] > 1 ,'Up','Down'),
'Stable')
#重命名
colnames(STADvol) <- c('log2FC','-log10(FDR)','Regulate')
#随机选取4个上调基因,4个下调基因
up4 <- STADvol[sample(which(STADvol$Regulate=='Up'),4),1:2]
down4 <- STADvol[sample(which(STADvol$Regulate=='Down'),4),1:2]
all8 <- rbind(up4,down4)画图:
#绘制火山图
library(ggplot2)
ggplot(
#设置数据
STADvol,
aes(x = log2FC, y = `-log10(FDR)`, colour=Regulate)) +
geom_point(alpha=0.4, size=1) +
scale_color_manual(values=c("#4393C3", "#d2dae2","#D6604D"))+
# 辅助线
geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +
geom_hline(yintercept = 2,lty=4,col="black",lwd=0.8) +
# 坐标轴
labs(x="log2(FC)",
y="-log10(FDR)")+
theme_bw()+
# 图例
theme(plot.title = element_text(hjust = 0.5),
legend.position="right",
legend.title = element_blank()
)+geom_text(data=all8,aes(label=rownames(all8)),color="black")#表示上调下调基因,可根据需求而定
结果展示:
三、功能富集分析
#功能富集分析
#载入相关包
library(clusterProfiler)
library(org.Hs.eg.db)
#提取上调基因和下调基因数据
up <- STADdata[which(STADvol$Regulate=='Up'),]
down <- STADdata[which(STADvol$Regulate=='Down'),]
#gene名转换
upgene <- bitr(rownames(up),fromType = "SYMBOL",toType = "ENTREZID",OrgDb = "org.Hs.eg.db")
downgene <- bitr(rownames(down),fromType = "SYMBOL",toType = "ENTREZID",OrgDb = "org.Hs.eg.db")
#上调基因功能富集
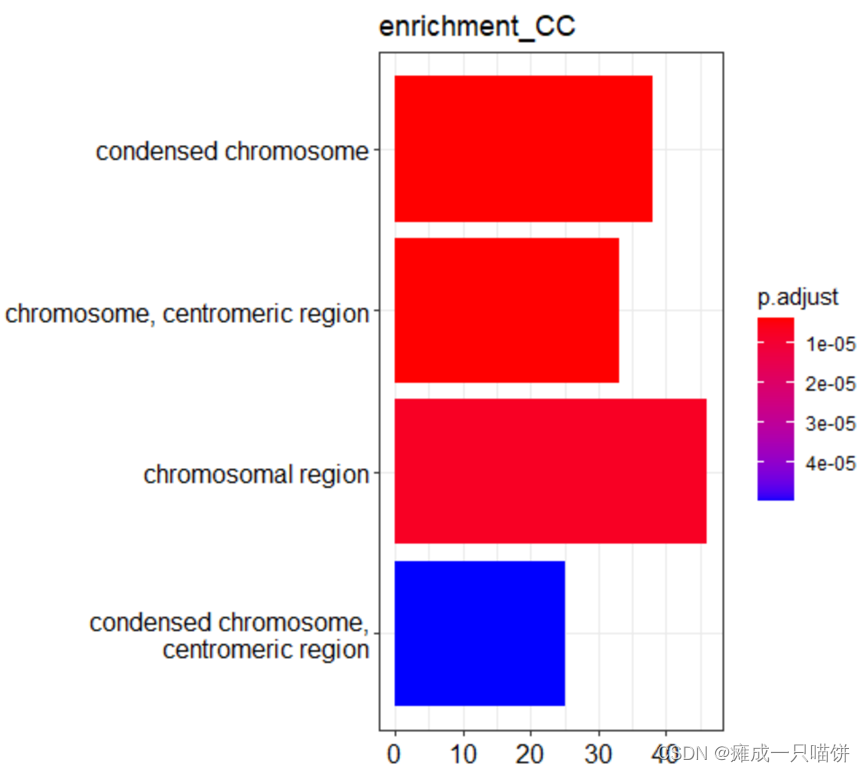
Go_CC <- enrichGO(gene = upgene[,2],
OrgDb=org.Hs.eg.db,
ont = "CC",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
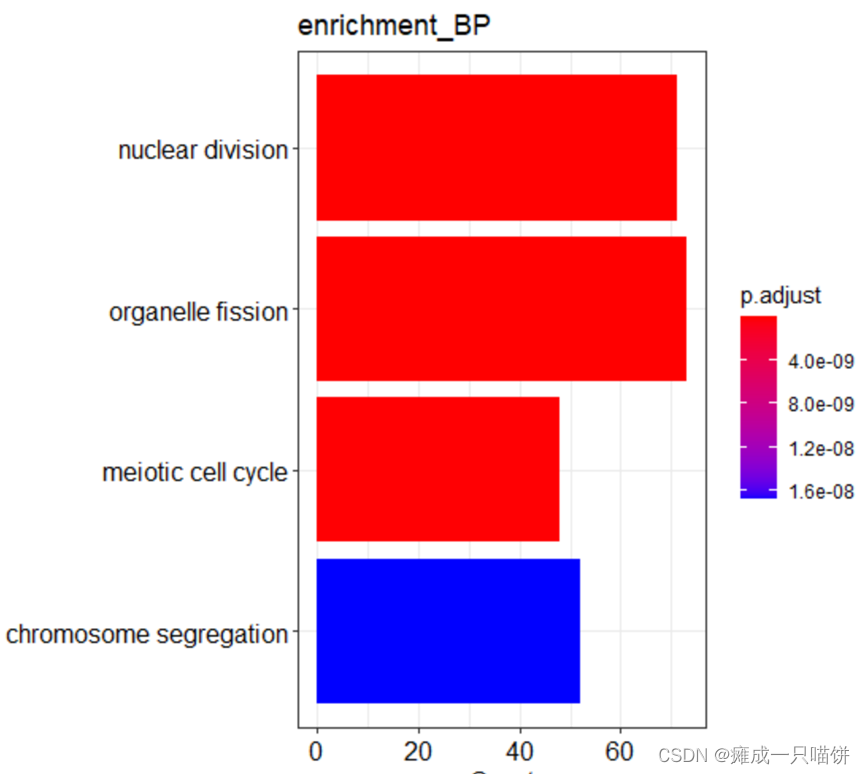
Go_BP <- enrichGO(gene = upgene[,2],
OrgDb=org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
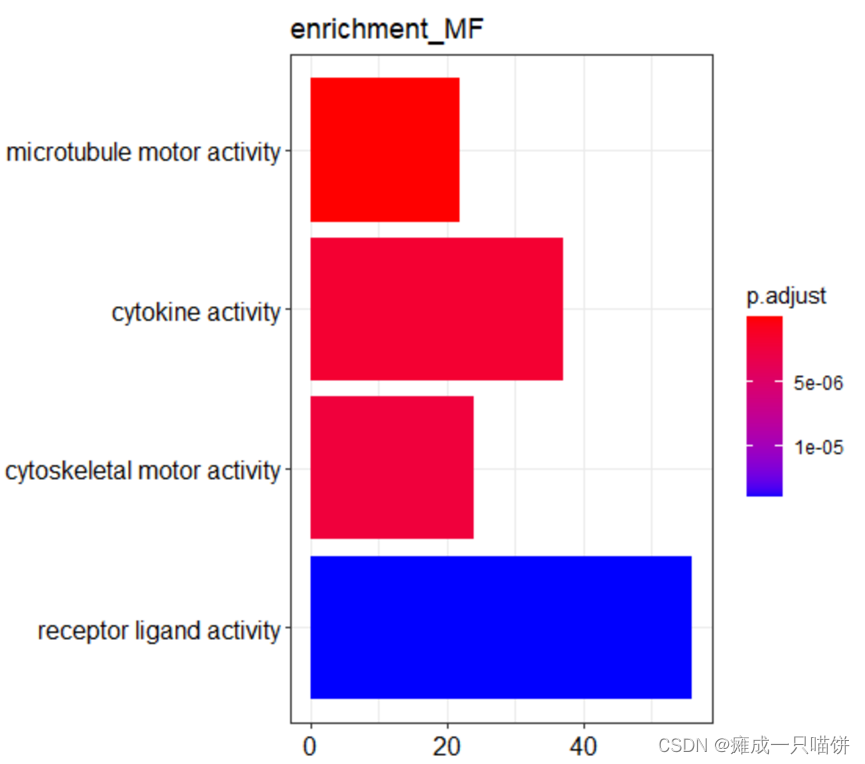
Go_MF <- enrichGO(gene = upgene[,2],
OrgDb=org.Hs.eg.db,
ont = "MF",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
#画图
barplot(Go_CC,drop = TRUE,title = "enrichment_CC",showCategory = 4)
barplot(Go_BP,drop = TRUE,title = "enrichment_BP",showCategory = 4)
barplot(Go_MF,drop = TRUE,title = "enrichment_MF",showCategory = 4)
#下调基因功能富集
Go_CC2 <- enrichGO(gene = downgene[,2],
OrgDb=org.Hs.eg.db,
ont = "CC",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
Go_BP2 <- enrichGO(gene = downgene[,2],
OrgDb=org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
Go_MF2 <- enrichGO(gene = downgene[,2],
OrgDb=org.Hs.eg.db,
ont = "MF",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
barplot(Go_CC2,drop = TRUE,title = "DownGene_enrichment_CC",showCategory = 4)
barplot(Go_BP2,drop = TRUE,title = "DownGene_enrichment_BP",showCategory = 4)
barplot(Go_MF2,drop = TRUE,title = "DownGene_enrichment_MF",showCategory = 4)
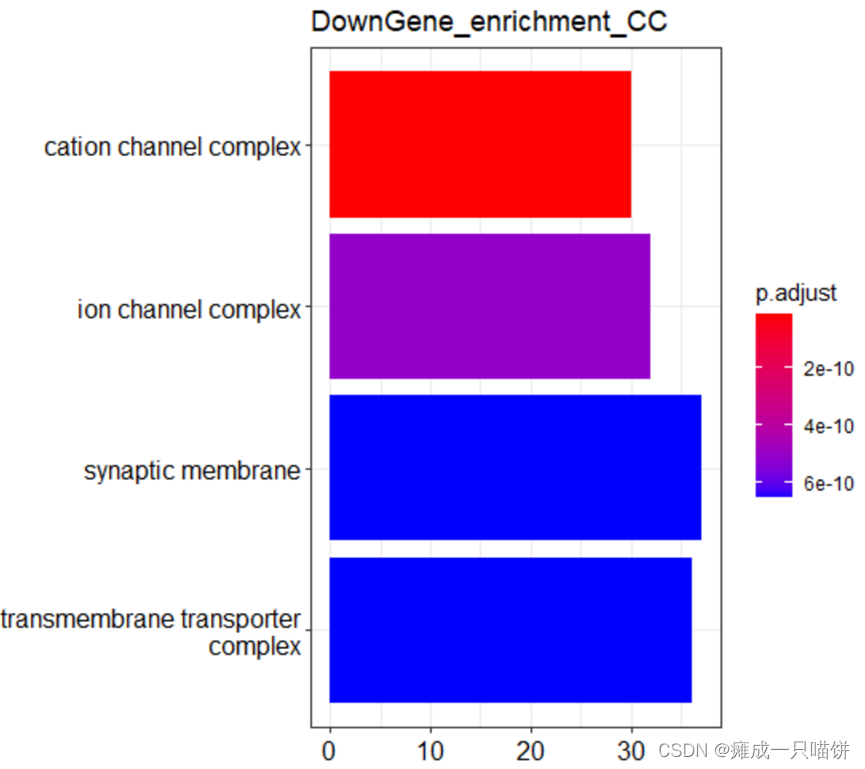
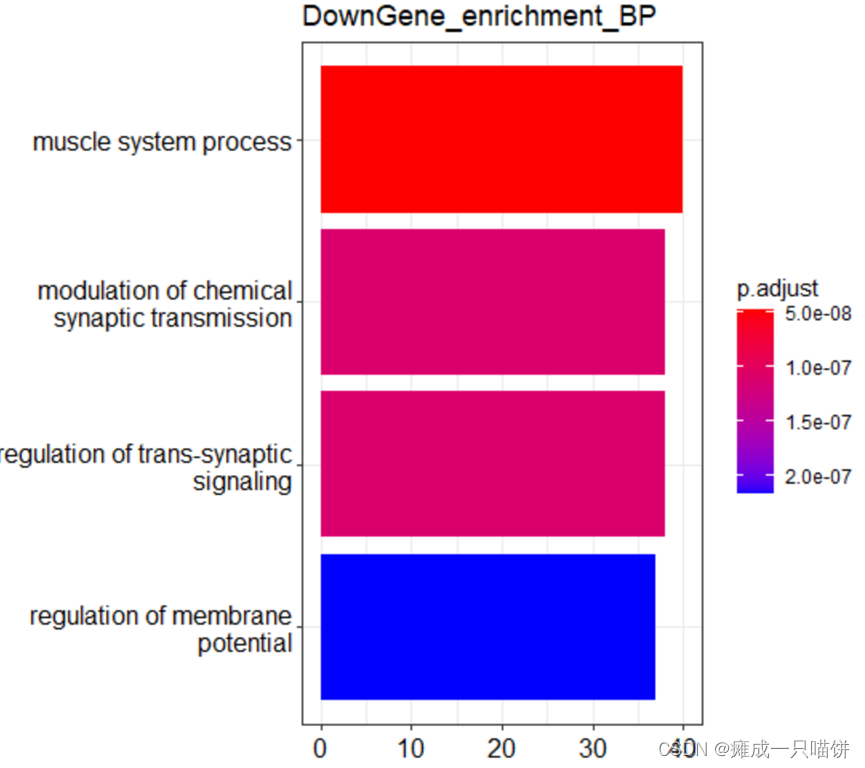
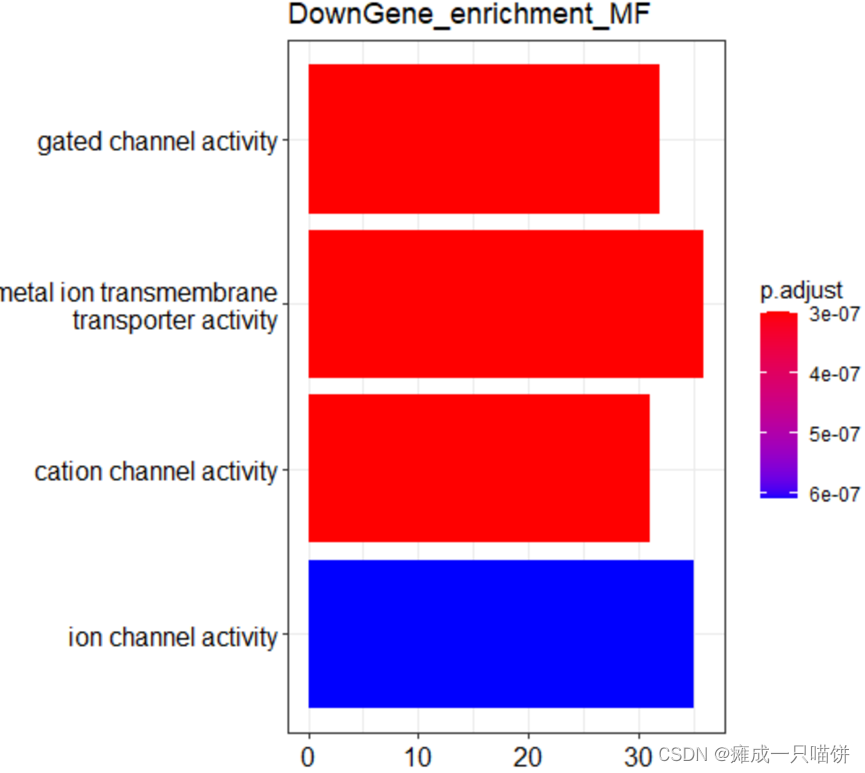
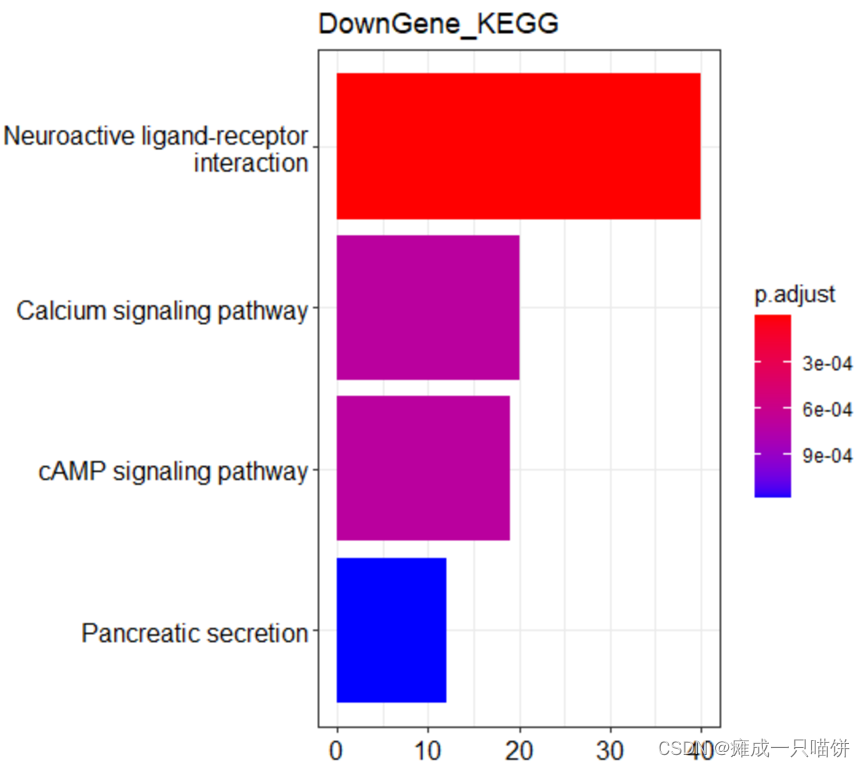
四、通路富集分析
#上调基因通路富集
upkk <- enrichKEGG(gene = upgene[,2],
organism ="human",
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
minGSSize = 1,
use_internal_data =FALSE)
#下调基因通路富集
downkk <- enrichKEGG(gene = downgene[,2],
organism ="human",
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
minGSSize = 1,
#readable = TRUE ,
use_internal_data =FALSE)
barplot(upkk,drop = TRUE,title = "UPGene_KEGG",showCategory = 4)
barplot(downkk,drop = TRUE,title = "DownGene_KEGG",showCategory = 4)
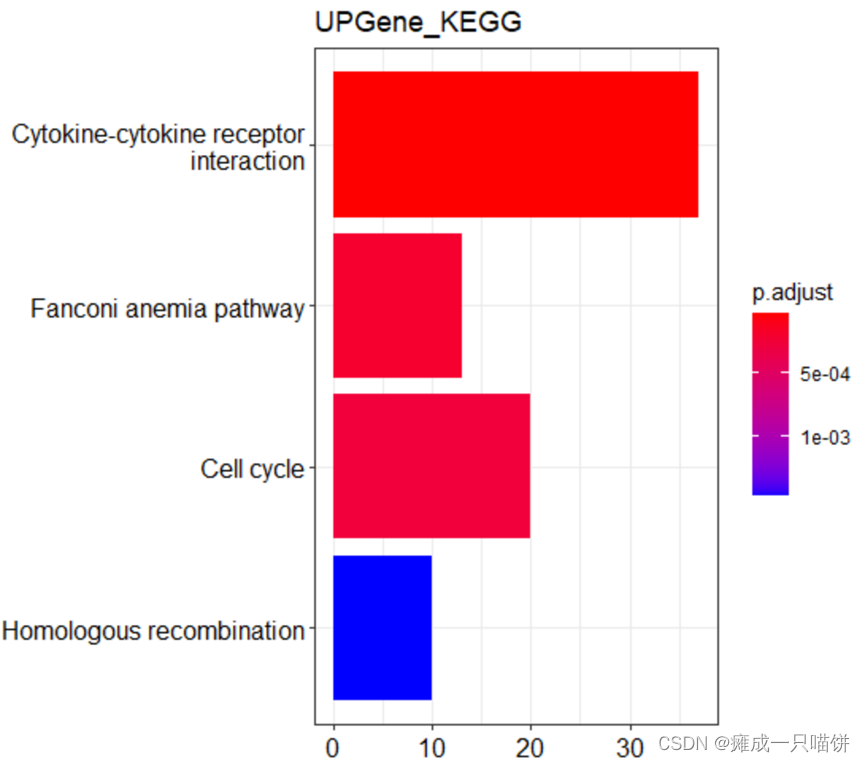
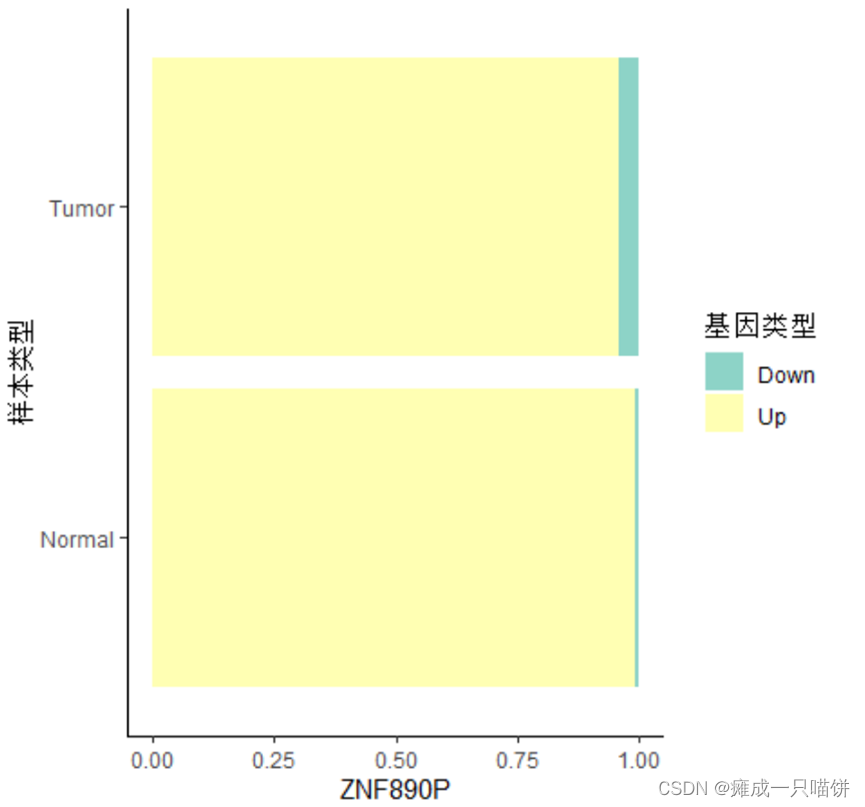
五、所挑选的上调基因与下调基因在癌症和正常样本中的比例
#载入相关包
library(ggplot2)
library(ggstatsplot)
#上调第一个基因与癌症中其他基因的相关性
up1cort <- apply(STADdata,1,function(x){
cor.test(as.numeric(STADdata[rownames(up4)[1],1:375]),
as.numeric(x[1:375]),method="pearson")$p.value
})
p1 <- table(STADvol[which(up1cort<0.01),3])
#上调第一个基因与正常中其他基因的相关性
up1corn <- apply(STADdata,1,function(x){
cor.test(as.numeric(STADdata[rownames(up4)[1],376:407]),
as.numeric(x[376:407]),method="pearson")$p.value
})
p2 <- table(STADvol[which(up1corn<0.01),3])
#数据整合
temp <- data.frame(t(rbind(c(p1[-2],p2[-2]),
c("Down","Up","Down","Up"),
c("Tumor","Tumor","Normal","Normal"))))
class1 <- apply(temp, 1, function(x){rep(x[2],as.numeric(x[1]))})
class2 <- apply(temp, 1, function(x){rep(x[3],as.numeric(x[1]))})
prodata <- data.frame(cbind(unlist(class1),unlist(class2)))
colnames(prodata) <- c("基因类型","样本类型")
#画图
prodata %>%
ggplot(aes(x =样本类型, fill =基因类型)) +
geom_bar(position = position_fill()) +
scale_fill_brewer(palette = 'Set3') + #设置颜色板
theme_classic() + #设置主题
labs(y = rownames(up4)[1]) + #子标题为基因名
coord_flip() #旋转坐标轴
#下面同理
#上调第2个基因与癌症中其他基因的相关性
up2cort <- apply(STADdata,1,function(x){
cor.test(as.numeric(STADdata[rownames(up4)[2],1:375]),
as.numeric(x[1:375]),method="pearson")$p.value
})
p1 <- table(STADvol[which(up2cort<0.01),3])
#上调第2个基因与正常中其他基因的相关性
up2corn <- apply(STADdata,1,function(x){
cor.test(as.numeric(STADdata[rownames(up4)[2],376:407]),
as.numeric(x[376:407]),method="pearson")$p.value
})
p2 <- table(STADvol[which(up2corn<0.01),3])
#数据整合
temp <- data.frame(t(rbind(c(p1[-2],p2[-2]),
c("Down","Up","Down","Up"),
c("Tumor","Tumor","Normal","Normal"))))
class1 <- apply(temp, 1, function(x){rep(x[2],as.numeric(x[1]))})
class2 <- apply(temp, 1, function(x){rep(x[3],as.numeric(x[1]))})
prodata <- data.frame(cbind(unlist(class1),unlist(class2)))
colnames(prodata) <- c("基因类型","样本类型")
#画图
prodata %>%
ggplot(aes(x =样本类型, fill =基因类型)) +
geom_bar(position = position_fill()) +
scale_fill_brewer(palette = 'Set3') +
theme_classic() +
labs(y = rownames(up4)[2]) +
coord_flip() #旋转坐标轴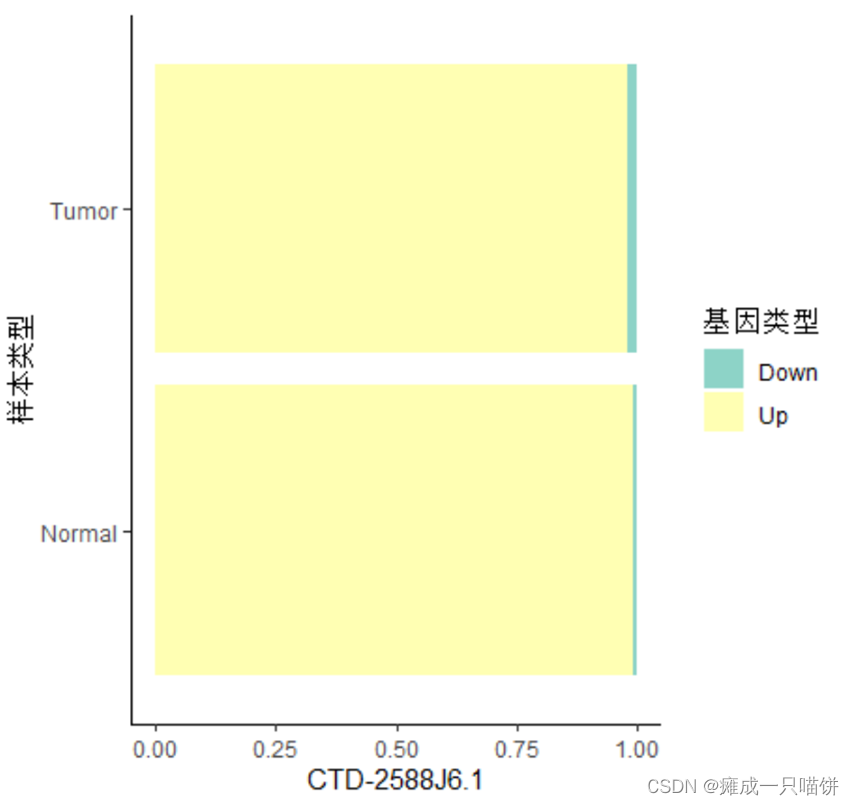
之后同理,可得剩下2个上调基因在差异基因集中Down与Up的比例:
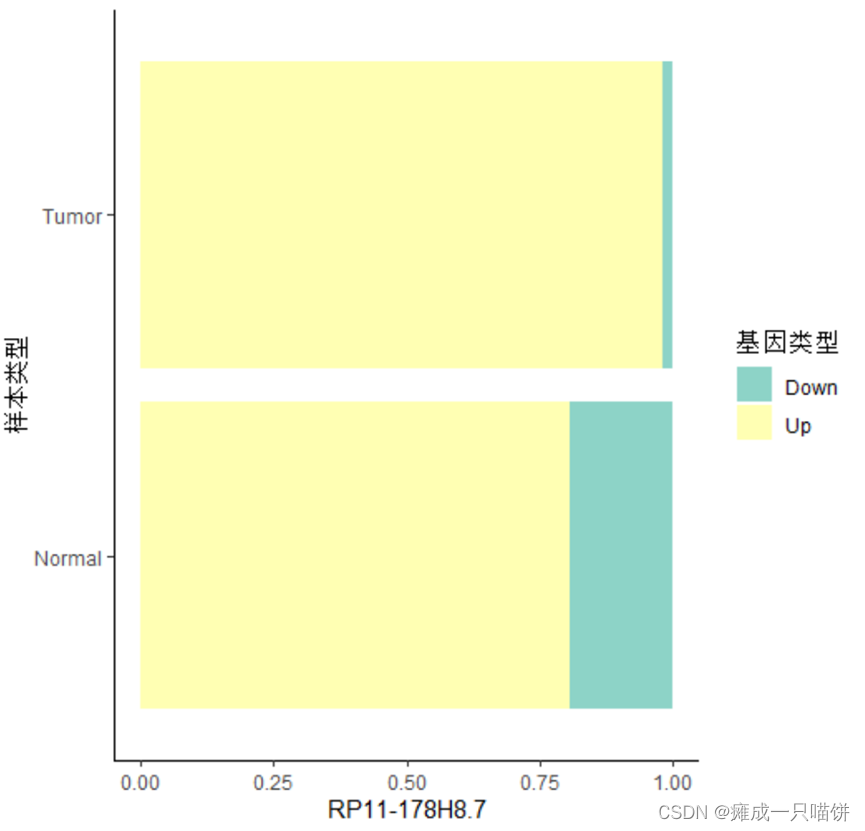
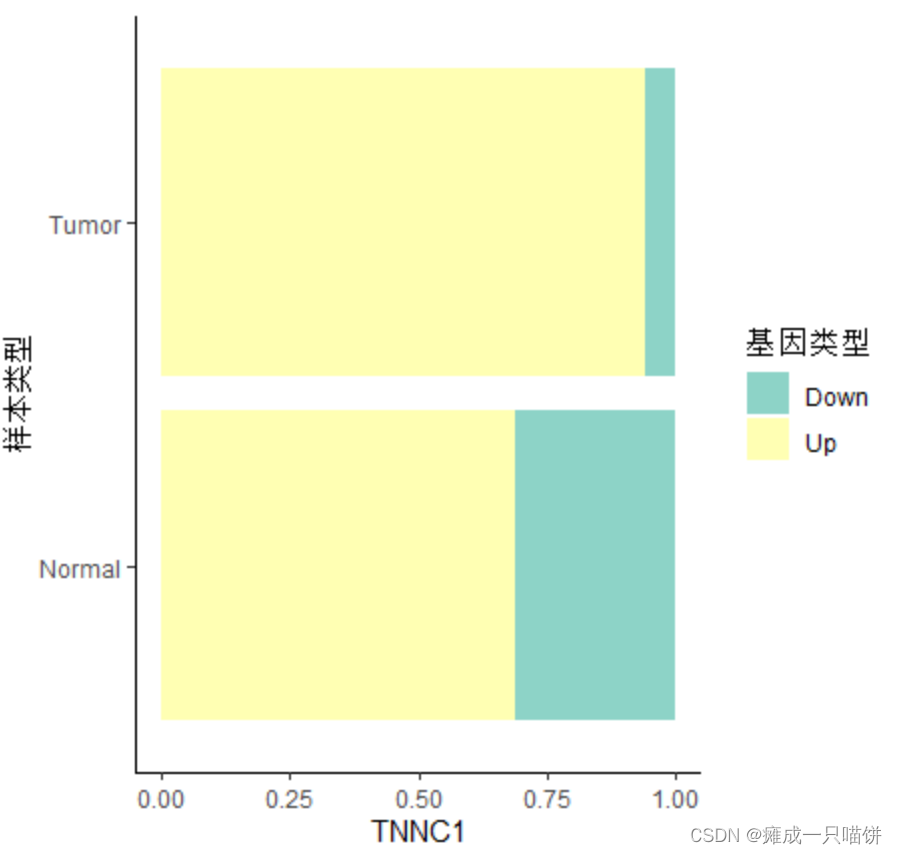
4个下调基因:
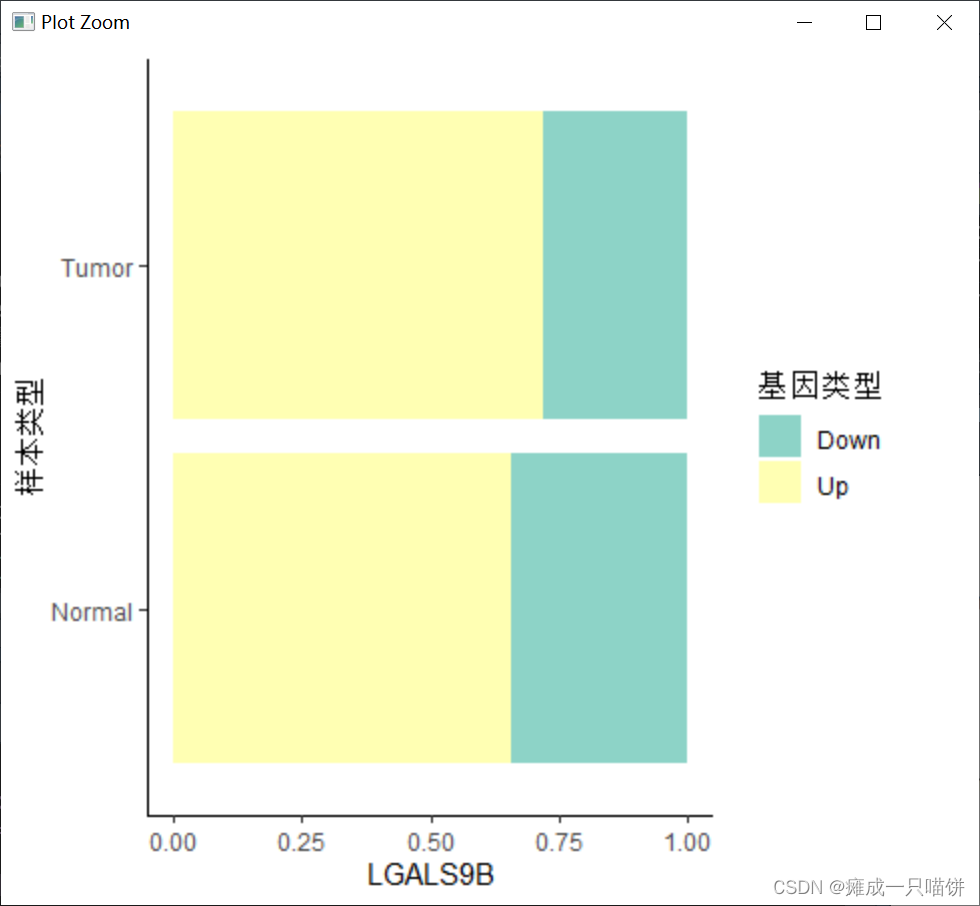
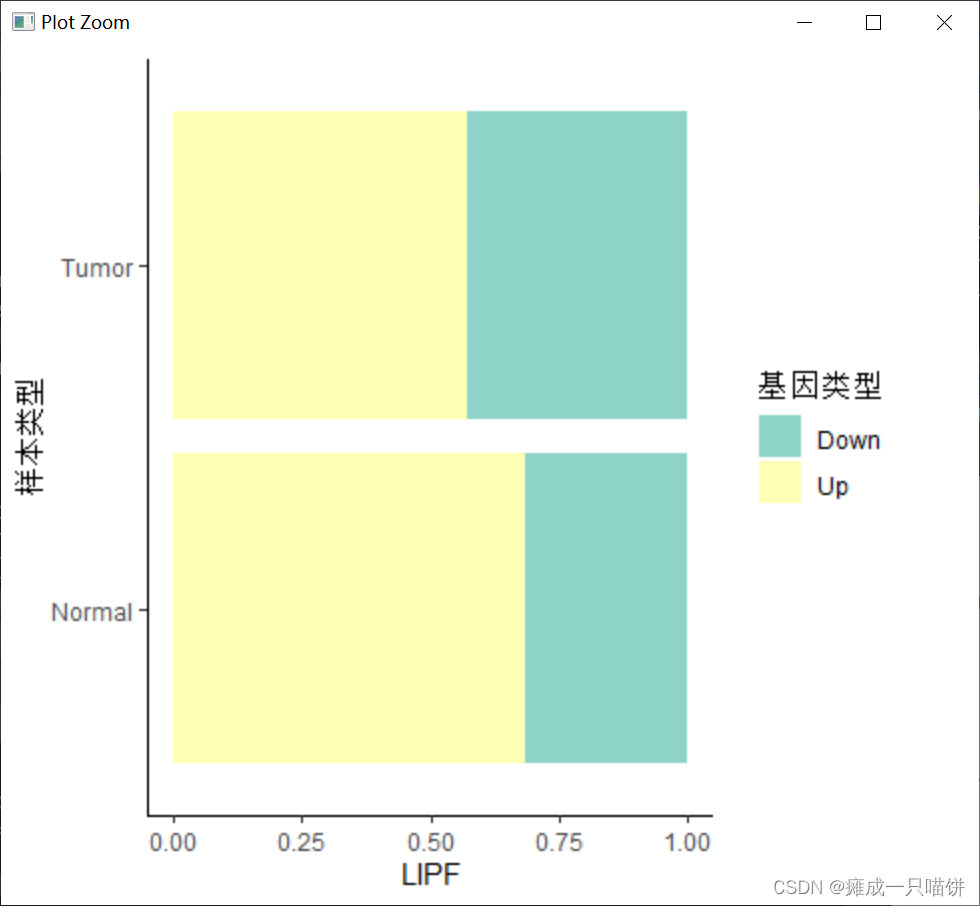
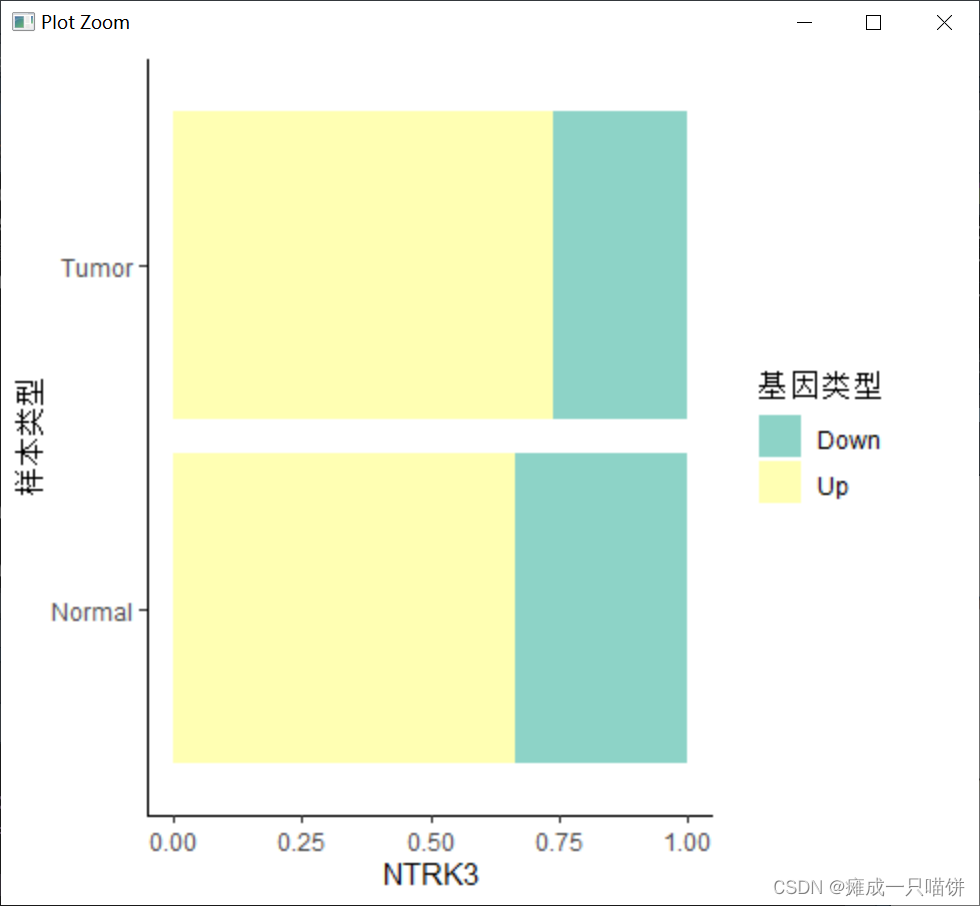
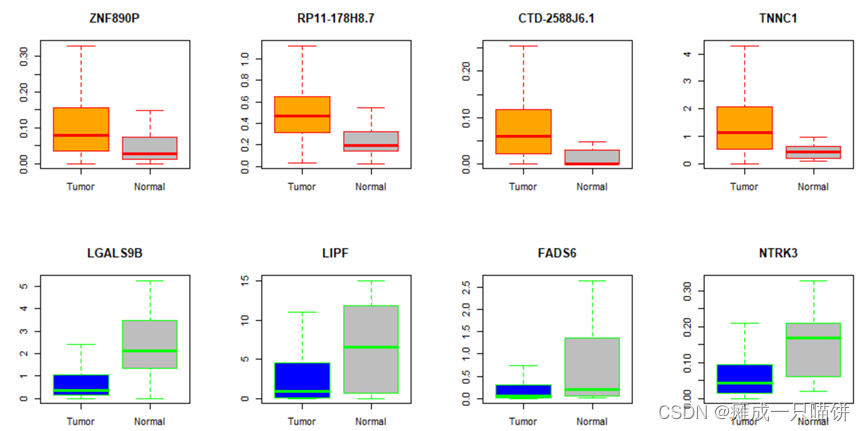
六、用箱式图表示上调和下调基因在癌症与正常样本中的表达
#数据准备
up4DataTu <- STADdata[rownames(up4),1:375]
up4DataNor <- STADdata[rownames(up4),376:407]
down4DataTu <- STADdata[rownames(down4),1:375]
down4DataNor <- STADdata[rownames(down4),376:407]
#画图
par(mfrow=c(2,4))
#上调第1个基因在癌症和正常样本中的表达
boxplot(t(up4DataTu)[,1],t(up4DataNor)[,1],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(up4DataTu)[1],
names = c("Tumor","Normal"),
col = c("orange","grey"),border = "red")
#上调第2个基因在癌症和正常样本中的表达
boxplot(t(up4DataTu)[,2],t(up4DataNor)[,2],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(up4DataTu)[2],
names = c("Tumor","Normal"),
col = c("orange","grey"),border = "red")
#上调第3个基因在癌症和正常样本中的表达
boxplot(t(up4DataTu)[,3],t(up4DataNor)[,3],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(up4DataTu)[3],
names = c("Tumor","Normal"),
col = c("orange","grey"),border = "red")
#上调第4个基因在癌症和正常样本中的表达
boxplot(t(up4DataTu)[,4],t(up4DataNor)[,4],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(up4DataTu)[4],
names = c("Tumor","Normal"),
col = c("orange","grey"),border = "red")
#下调第1个基因在癌症和正常样本中的表达
boxplot(t(down4DataTu)[,1],t(down4DataNor)[,1],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(down4DataTu)[1],
names = c("Tumor","Normal"),
col = c("blue","grey"),border = "green")
#下调第2个基因在癌症和正常样本中的表达
boxplot(t(down4DataTu)[,2],t(down4DataNor)[,2],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(down4DataTu)[2],
names = c("Tumor","Normal"),
col = c("blue","grey"),border = "green")
#下调第3个基因在癌症和正常样本中的表达
boxplot(t(down4DataTu)[,3],t(down4DataNor)[,3],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(down4DataTu)[3],
names = c("Tumor","Normal"),
col = c("blue","grey"),border = "green")
#下调第1个基因在癌症和正常样本中的表达
boxplot(t(down4DataTu)[,4],t(down4DataNor)[,4],xlim=c(0,2),at=c(0.5,1.5),
outline = F,main=rownames(down4DataTu)[4],
names = c("Tumor","Normal"),
col = c("blue","grey"),border = "green")
结果展示:

















 4098
4098

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









