








前情提要:算是补充基础知识记录
目录
直接上实战
1.for循环 和if语句
目的:把下列id根据11,12位的数字确定样本位置;即小于等于10为tumor,大于10为normal
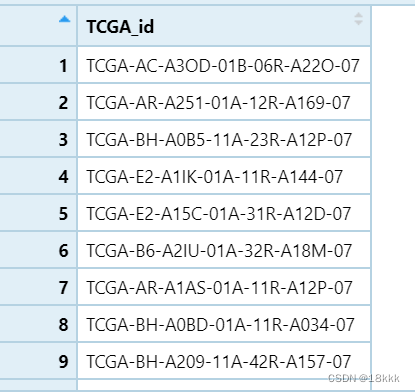
答案对照:
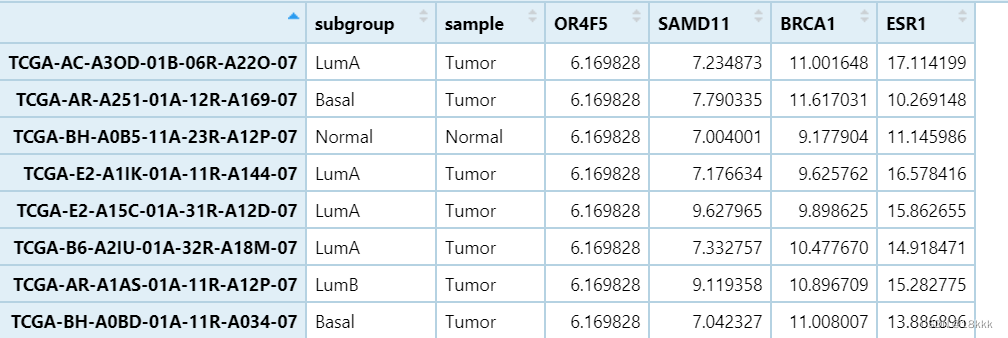
代码一
for (i in 1:length(metadata$TCGA)) {
ifelse(substring(metadata$TCGA_id[[i]],14,15)<=10,
metadata$tissue[[i]] <- "Tumor",
metadata$tissue[[i]] <- "Normal")
}代码二
for (i in 1:length(metadata$TCGA)) {
if (substring(metadata$TCGA_id[[i]],14,15)<=10) {
metadata$tissue[[i]] <- "Tumor"
}
else{
metadata$tissue[[i]] <- "Normal"
}
}结果:
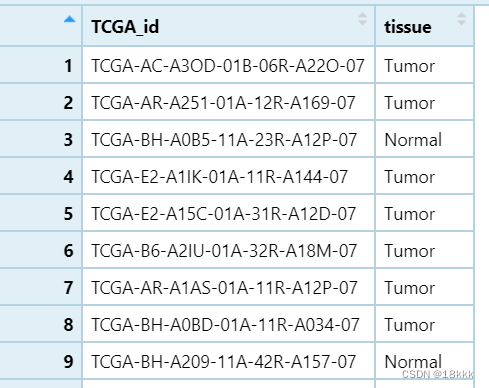
代码三果子
metadata <- data.frame(TCGA_id=rownames(exprSet))
### 1.首先确定框架,我们想重复的操作是什么?
### 2.单次操作应该如何做
### 3.如何用R语言实现单次的需求
substring(metadata[1,1],14,15)
##########################
### 批量操作的具体实现过程:
### 1.创建容器
metadata <- data.frame(TCGA_id=rownames(exprSet))
### 2.循环输出内容到容器
for (i in 1:nrow(metadata)) {
## 指示
print(i)
## substring的用法,这是元素获取
num = as.numeric(substring(metadata[i,1],14,15))
#如果是肿瘤,就给第2列加上Tumor
if (num %in% seq(1,9)) {
metadata[i,2] = "Tumor"
}
#如果是正常组织,就给第2列加上Normal
if (num %in% seq(10,29)) {
metadata[i,2] = "Normal"
}
}
## 修改名称
colnames(metadata) <- c("TCGA_id","sample")
总结:
代码一和二是我写的,果子的代码看似复杂,但更有条理性
1.注意了字符串和数值的区别
我缺少as.numeric也可以运行,但忽视这种差异吃枣药丸

比如在我和答案比较时就会发现

这实际上是因为exprSet$sample是因子状态,所以使用identical为F,而直接比较大小则为T
2.ifelse
十分重要的函数
3.创造容器
做循环前一定要有容器,空的也可以
2.批量做基因的相关性分析
查看数据
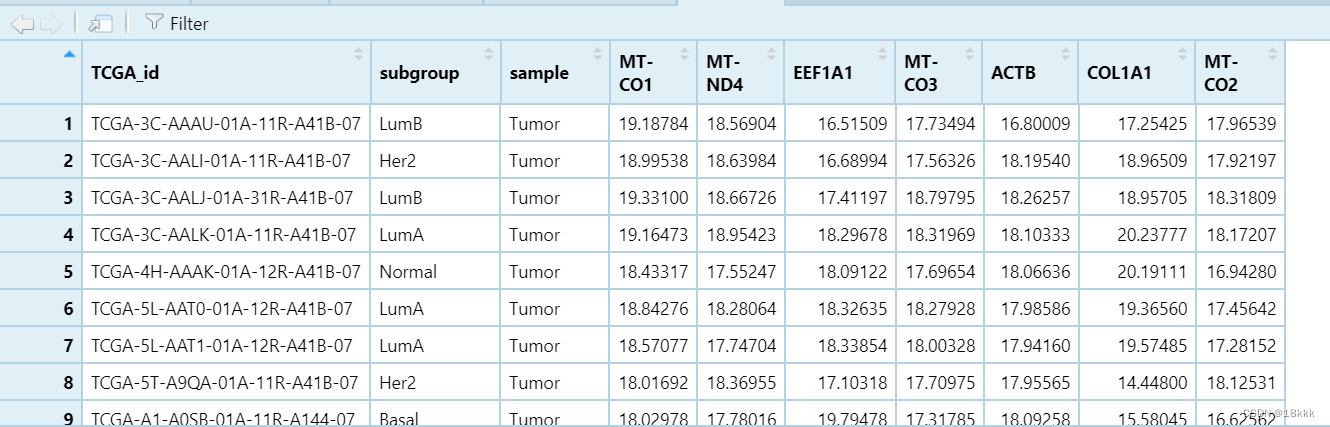
目的:做两个基因的相关性
因为我的思路一开始和果子的思路不太一样,就先介绍他的代码
代码一果子
为了简化,我对数据精简,只留下十个基因作为示例
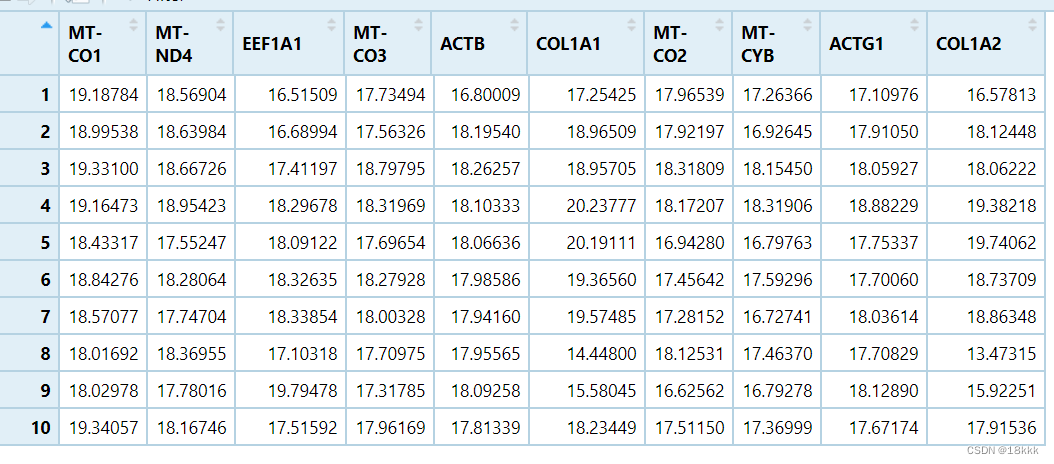
###果子
##1.框
correlation <- data.frame()
##2.修剪要循环的数据
data <- exprSet[,-c(1,2,3)]
test <- data[1:10,1:10]
##3.获取批量操作的范围,应该是个向量;以10个基因的test代替data
genelist <- colnames(test)
##4.开始for循环,数据导出到容器
gene <- "COL1A2"
genedata <- as.numeric(test[,gene])
for(i in 1:length(genelist)){
## 1.指示
print(i)
## 2.计算
dd = cor.test(genedata,as.numeric(test[,i]),method="spearman")
## 3.填充
correlation[i,1] = gene
correlation[i,2] = genelist[i]
correlation[i,3] = dd$estimate
correlation[i,4] = dd$p.value
}
colnames(correlation) <- c("gene1","gene2","cor","p.value")
结果:
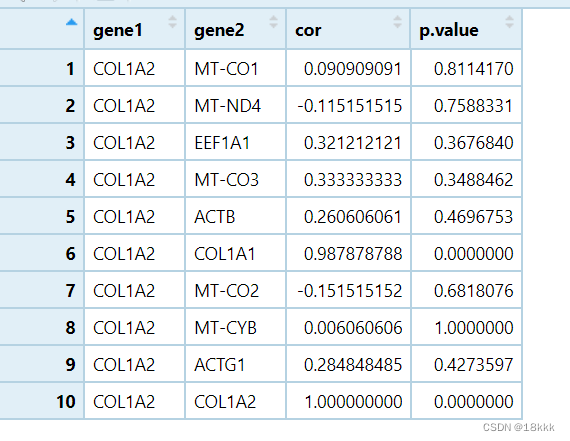
做一个基因和其它基因的相关性分析,并且导出p和相关性系数
代码二:我的思路
我的想法是,在做两样本的相关性分析/两细胞群的相关性分析时,我们一般把数据排列成行为基因名,列为样本名的形式,做cor()函数即可得到相关的矩阵;现在做两基因的相关性分析,把基因放在列名就可以了呀。正好和函数的样式相一致。
于是我用
genecor <-cor(test, method='spearman') 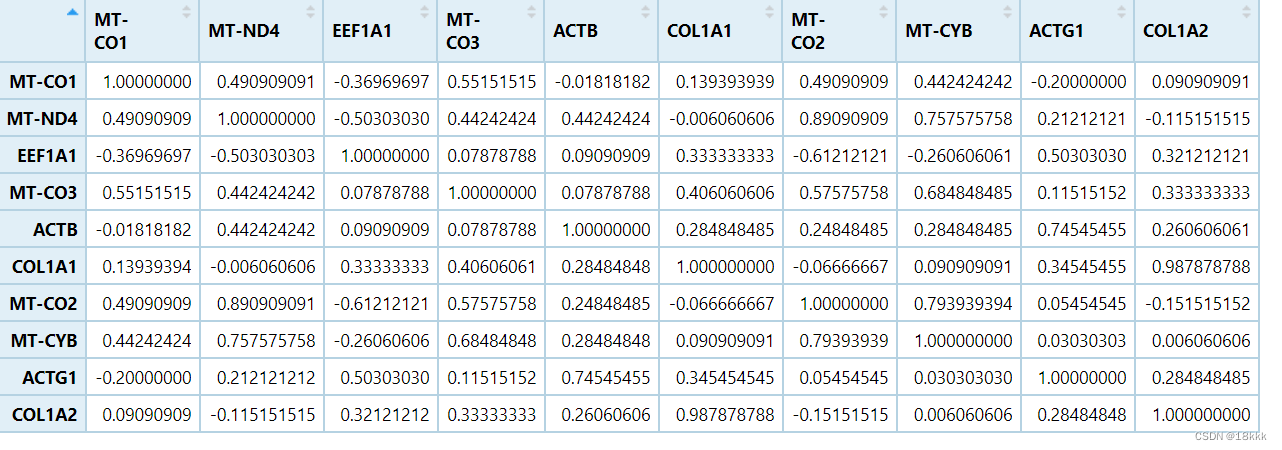
比较一下和果子的结果

不太懂为什么最后一个是F,但反正可以说两个结果是一致的,但是单一的cor函数难以给出p值
总结
cor()
cor.test()
是两个不同的函数,cor.test可以提供更多的信息
更多情况下可以直接用一些R包绘制相关性的cor_plot
贴两个学习路径
单基因批量相关性分析的妙用
## https://mp.weixin.qq.com/s/TfE2koPhSkFxTWpb7TlGKA
## 单基因批量相关性分析的GSEA
## https://mp.weixin.qq.com/s/sZJPW8OWaLNBiXXrs7UYFw
如需测试数据,请发邮件至yunbk@mail2.sysu.edu.cn



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











