









今天来读取和下载一份单细胞数据
思路来自上一篇文章的学习http://t.csdn.cn/qR4zv
GSE139324
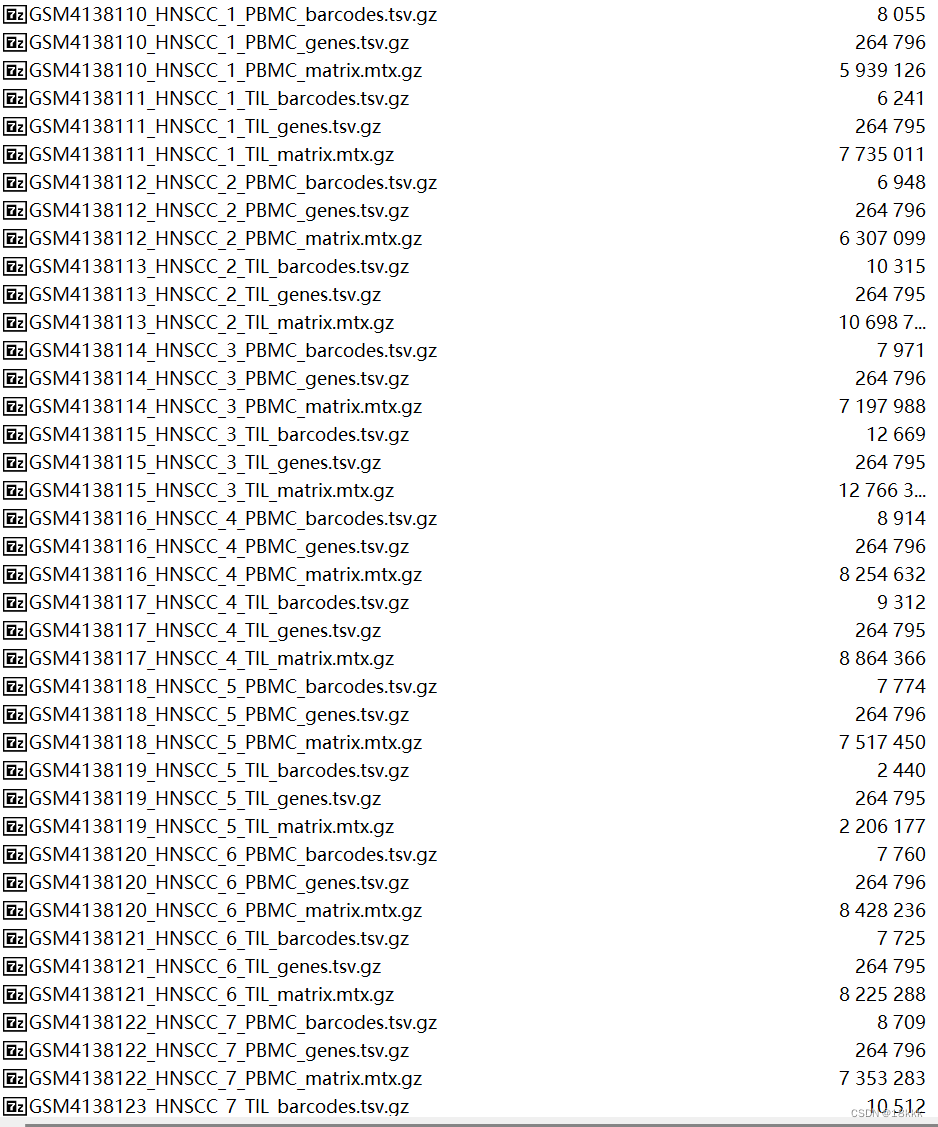
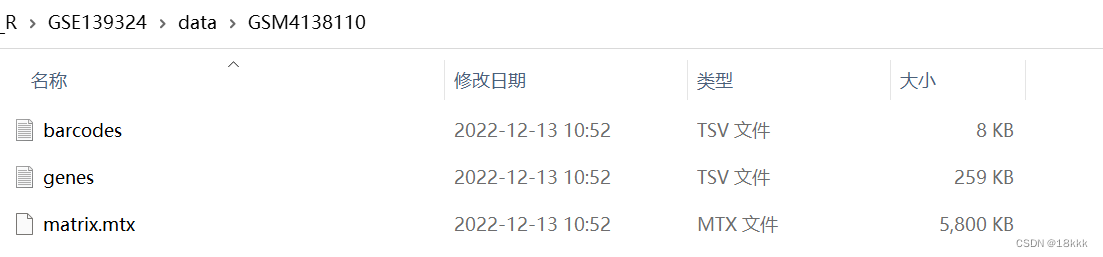
下载好以后发现数据是这样的,我们最终的目的应该是这样的:三个文件依次放在sample名下
之前都是手动修改,这次我们用R语言来修改和读取
1.用getGEO读取一下临床信息
library(GEOquery)
if(T){
eSet <- getGEO('GSE139324',
AnnotGPL = F,
getGPL = T)
eSet$GSE164690_series_matrix.txt.gz@phenoData
pd <- Biobase::pData(eSet[[1]]) # 临床信息
dat <- Biobase::exprs(eSet[[1]]) # 表达矩阵
save(pd,file="results/getGEO_GSE164690.rda")
}pd有点鸡肋
2.整理scRNAdata
把上述RAW文件夹放在GSE139324的目录下
rm(list = ls())
if(!file.exists("GSE139324/data")) dir.create("GSE139324/data", recursive = T)
data_path <- "GSE139324/data/"
files <- list.files("GSE139324/GSE139324_RAW/");files
###GSE名称
GSE <- unique(str_split(files,"_",simplify = T)[,1]);GSE
3.建立GSE的空文件夹
for (i in GSE) {
dir.create(paste0(data_path,i))
}
4. 批量处理数据
for (i in 1:length(GSE)) {
print(i)
myfile <- paste0(getwd(),"/GSE139324/GSE139324_RAW/",files[str_detect(files,GSE[[i]])])
file.copy(myfile,paste0(data_path,GSE[[i]]))
old_names <- list.files(paste0(data_path,GSE[[i]]),full.names = T)
new_names <- unique(str_split(old_names,"_",simplify = T))[,5]
new_names2 <-paste0(paste0(data_path,GSE[[i]]),"/",gsub(".gz","",new_names))
file.rename(old_names,new_names2)
}其实应该先写好一个数据,随后写入循环就行了,一个样本的处理如下:
###先做一个
myfile <- paste0(getwd(),"/GSE139324/GSE139324_RAW/",files[str_detect(files,GSE[[1]])]);myfile
file.copy(myfile,paste0(data_path,GSE[[1]]))
old_names <- list.files(paste0(data_path,GSE[[1]]),full.names = T)
new_names <- unique(str_split(old_names,"_",simplify = T))[,5]
new_names2 <-paste0(paste0(data_path,GSE[[1]]),"/",gsub(".gz","",new_names))
file.rename(old_names,new_names2)首先按照样本名找到对应的三个文件

然后file.copy把他们拷贝入对应的GSE文件夹下
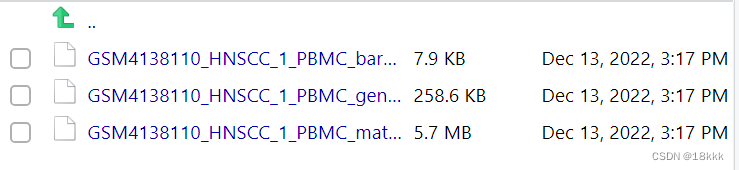
获取这三个文件的名称oldnames
 改新名字
改新名字
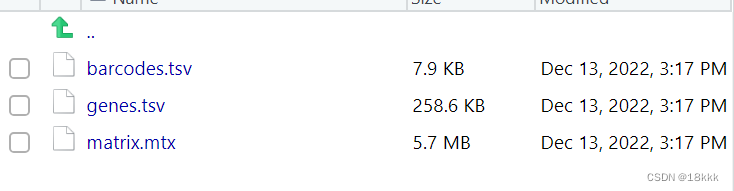
 完成名字修改file.rename(old_names,new_names2)
完成名字修改file.rename(old_names,new_names2)
5.读取数据
system.time({
sceList = lapply(GSE_files,function(patient){
# patient=files[[1]]
print(patient)
ct <- Read10X(patient)
sce=CreateSeuratObject(counts = ct ,
project = str_split_fixed(patient,"/",n=3)[,3],###即样本的GSM编号
min.cells = 3, #Include features detected in at least this many cells.
min.features = 200 # Include cells where at least this many features are detected.
)
return(sce)
}) #返回一个List
})#记录一下运行时间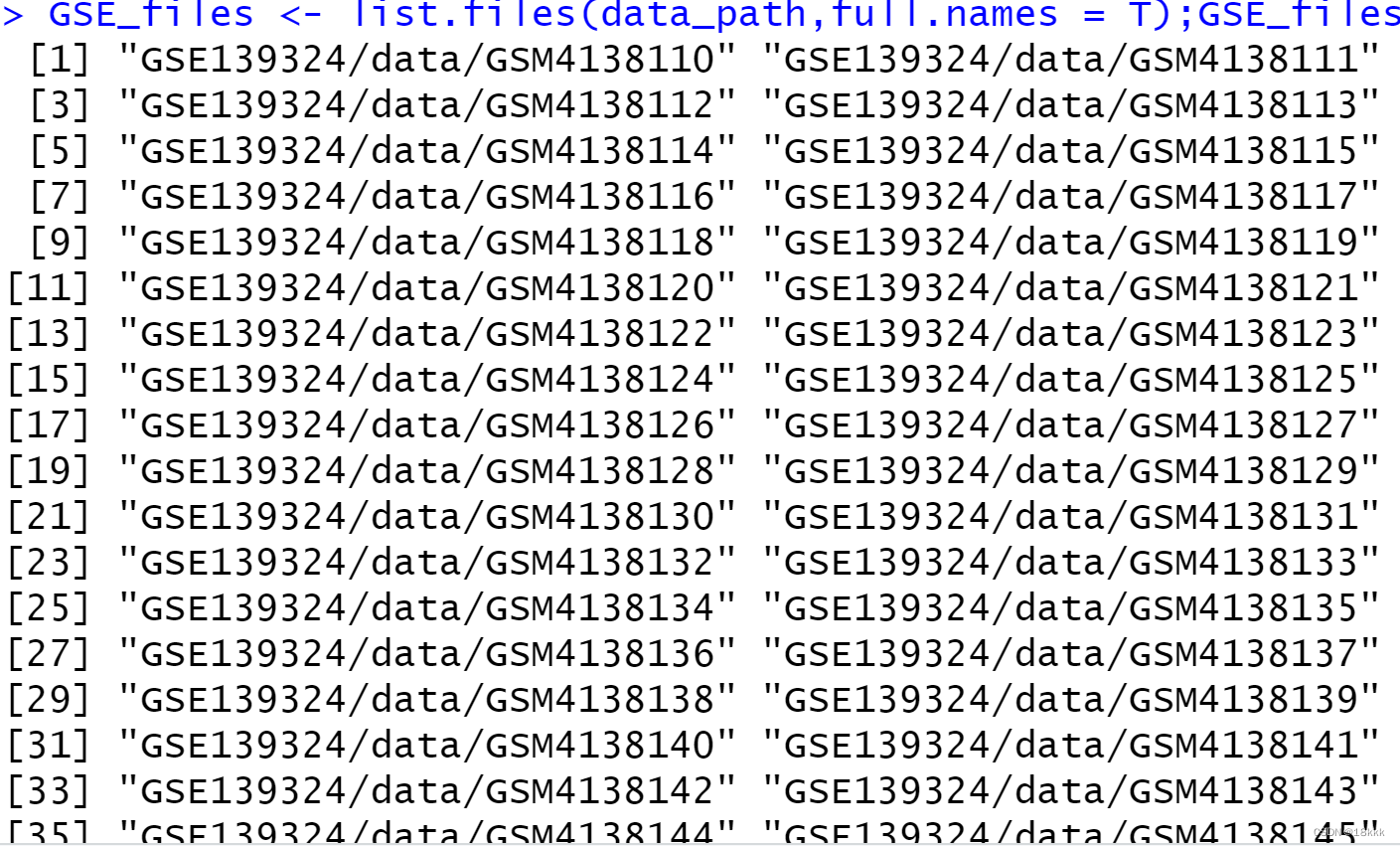
6.合并数据
names(sceList)
GSE
names(sceList) = GSE
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],add.cell.ids = GSE)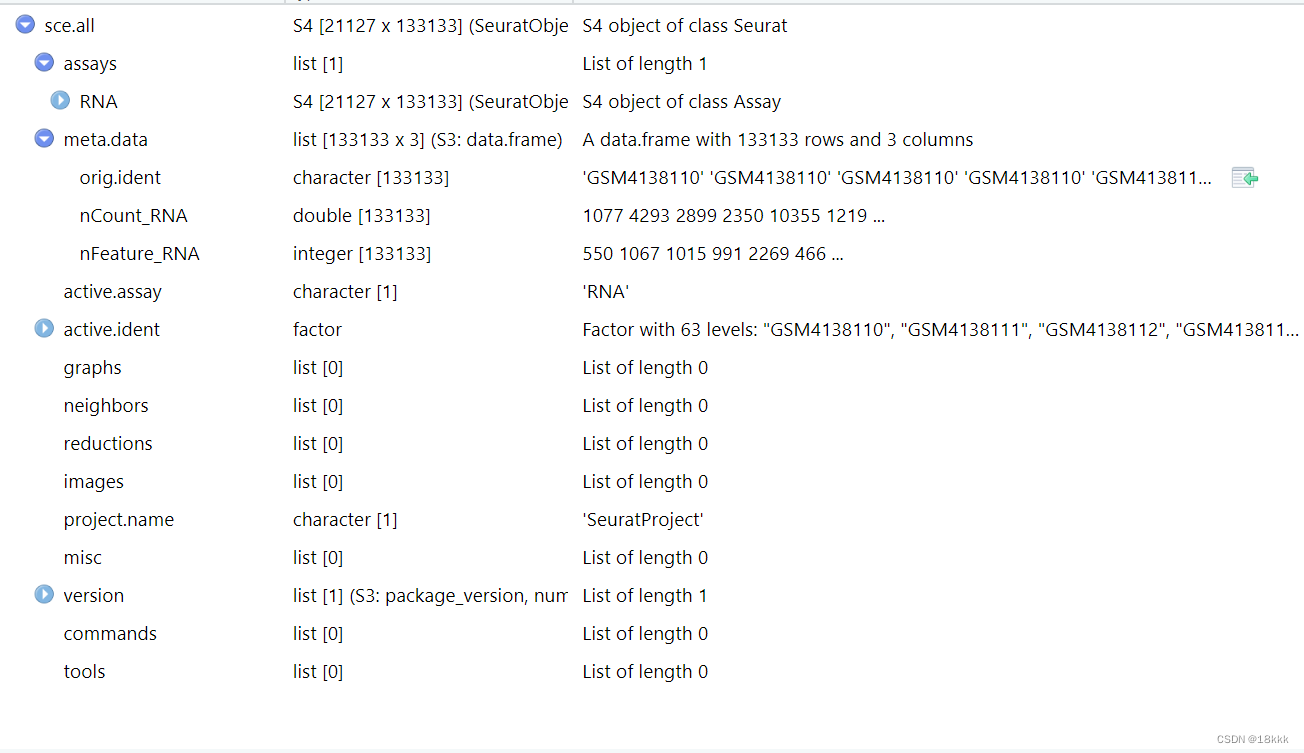
每个样本的细胞不算很多
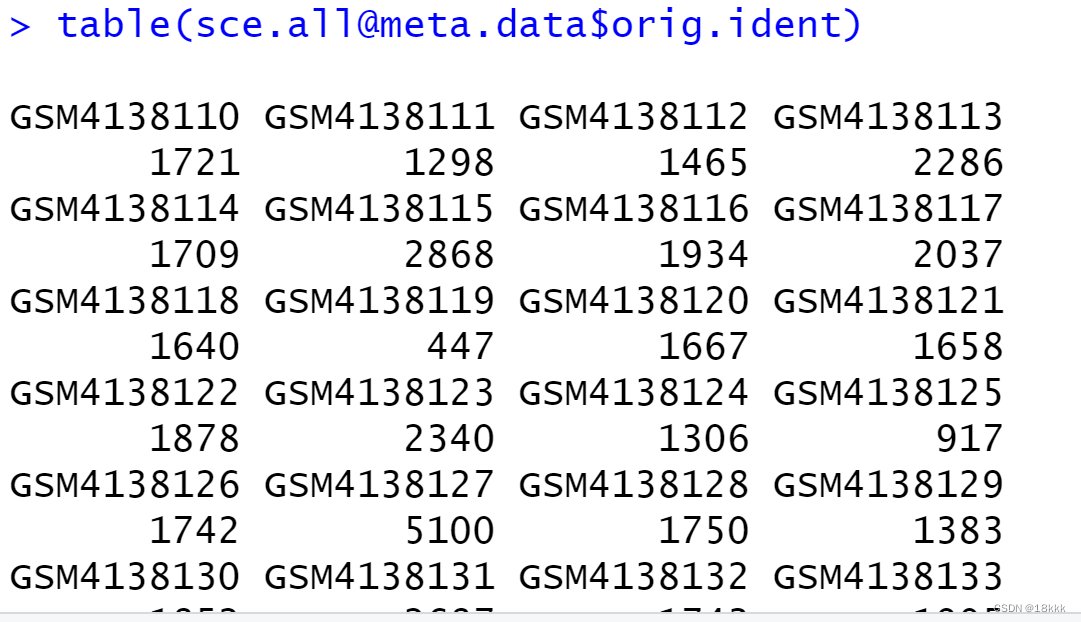
2023.06.06更新分界线
有时候会有些不规则的数据 比如下图这个,整个GSE20多个样本就给了一份数据,而且genes barcodes是txt格式的,正常的10X是使用tsv格式的,这样就只能分开读取

代码:
###分别读取数据
###矩阵必须用readMM读取稀疏矩阵
g1 <- fread('sc-RNA-raw-data/genes.txt.gz',data.table = F,header = F)
b1 <- fread('sc-RNA-raw-data/barcodes.txt.gz',data.table = F,header = F)
m1<-Matrix::readMM('sc-RNA-raw-data/matrix.mtx.gz')
dim(m1)
expr <-as.data.frame(m1)
test <- expr[1:5,1:5]
###看到基因名当中有重复的
head(sort(table(g1$V1),decreasing = T),50)
# 把对应的重复值给去掉 因为数量不多 直接去重复
# 同时还要对expr矩阵也做相同的操作
g2 <- g1[!duplicated(g1$V1),]
expr <- expr[!duplicated(g1$V1),]
###构建完整表达矩阵
rownames(expr) <- g2
colnames(expr) <- b1$V1
###seurat data
sce=CreateSeuratObject(counts = as.matrix(expr),
min.cells = 20, #文章OPSCC阈值
min.features = 1000,)#文章OPSCC阈值
saveRDS(sce,file = "sc-RNA-raw-data/raw-seurat-data.rds")
sce <- readRDS('sc-RNA-raw-data/raw-seurat-data.rds')
table(sce@meta.data$orig.ident)
看一下结果
虽然所有样本混在一起,但是创建Seurat对象的时候仍然可以识别来源
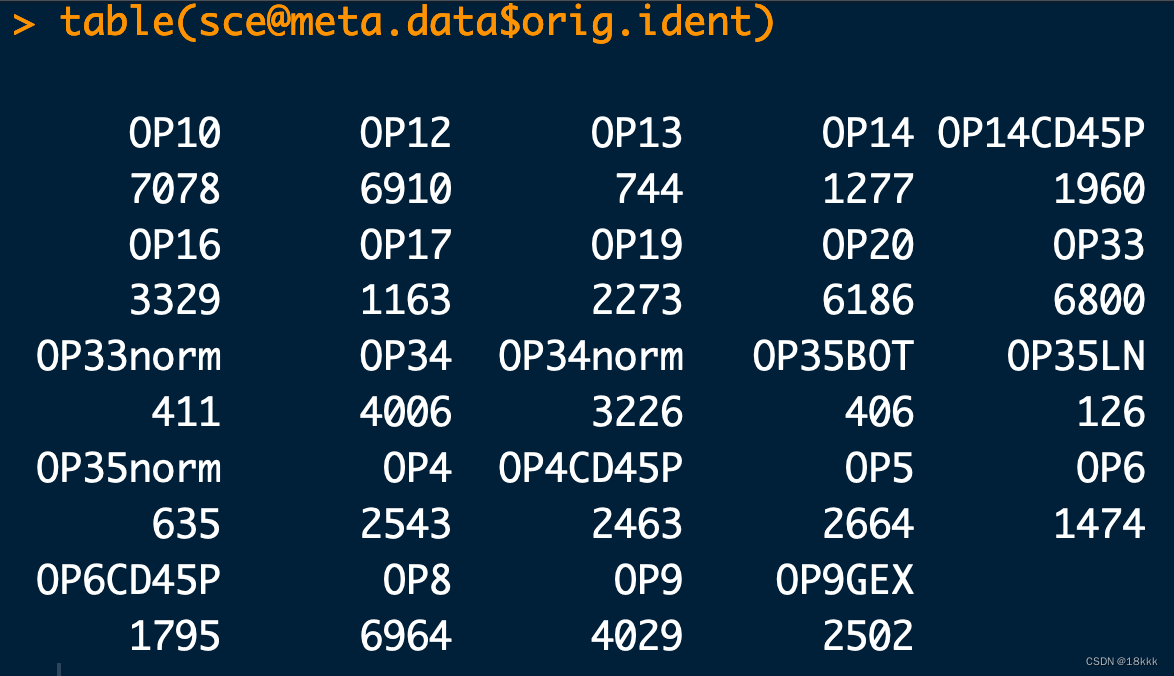
这是因为原来作者把barcodes带上了样本的ID,如果没有带的话在创建对象的时候要加上参数
> head(b1)
V1
1 OP10_AAACCCAAGAACGTGC-1
2 OP10_AAACCCAAGATGCGAC-1
3 OP10_AAACCCAAGCCATGCC-1
4 OP10_AAACCCAAGGAGCAAA-1
5 OP10_AAACCCACAAATAAGC-1
6 OP10_AAACCCACAACCAGAG-1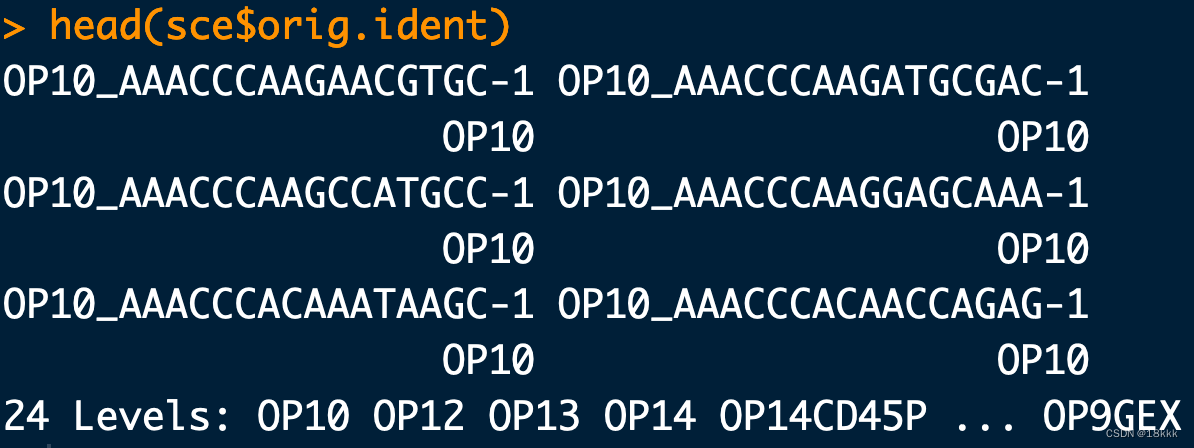



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











