







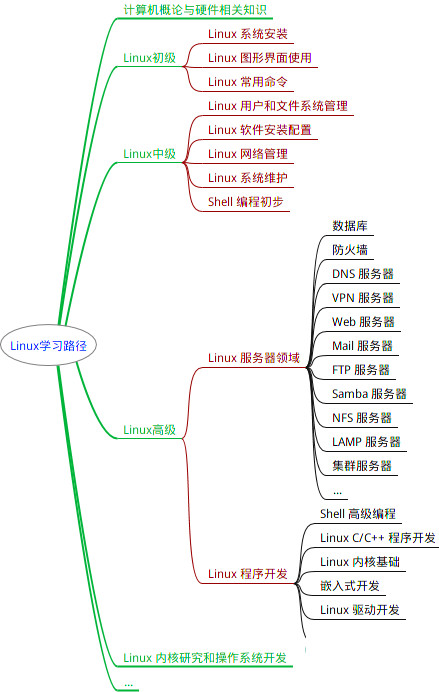
最全的Linux教程,Linux从入门到精通
======================
-
linux从入门到精通(第2版)
-
Linux系统移植
-
Linux驱动开发入门与实战
-
LINUX 系统移植 第2版
-
Linux开源网络全栈详解 从DPDK到OpenFlow

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
双端序列导入(序列文件与barcode文件各自独立)

获取样例文件
cd emp-paired-end-sequences
# 下载正向序列
wget \
-O forward.fastq.gz \
https://data.qiime2.org/2023.9/tutorials/atacama-soils/1p/forward.fastq.gz
#下载反向序列
wget \
-O reverse.fastq.gz \
https://data.qiime2.org/2023.9/tutorials/atacama-soils/1p/reverse.fastq.gz
# 下载barcodes文件
wget \
-O barcodes.fastq.gz \
https://data.qiime2.org/2023.9/tutorials/atacama-soils/1p/barcodes.fastq.gz
# 查看输入文件格式
# 正向序列文件内容
zcat forward.fastq.gz | head -n 10
@M00176:65:000000000-A41FR:1:1101:14282:1412 1:N:0:0
NACGTAGGGTGCAAGCGTTAATCGGAATTACNGGNNNTAAAGCGTGCNNAGGCNNNNNNNNNNNANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
#>>>AAF@ACAA4BGCEEECGGHGGEFCFBG#BA###BABAEFGEEE##BBAA###########B######################################################################################
@M00176:65:000000000-A41FR:1:1101:16939:1420 1:N:0:0
NACGTAGGGGGCAAGCGTTGTCCGGAATCATTGGNNGTAAAGAGCGTGNAGGCNNNNNGNNANNTNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
#>>ABAF@ABBBGGGGEEFGGGHGGFFGFHHHHH##BBEFDHHCGGFG#ABFF#####B##B##B######################################################################################
@M00176:65:000000000-A41FR:1:1101:14746:1560 1:N:0:0
TACGTAGGGAGCTAGCGTTGTCCGGAATCATTGGGCGTAAAGCGCGCGTAGGCGGCCAGATAAGTCCGGTGTAAAAGCCACAGGCTNNNNNNNNNNNNNNNNCNGGANNNNNNNNNNNNNNNNNNNNNNANNNNNNNNNNNNANNNNNGGN
# 反向序列文件内容
zcat reverse.fastq.gz | head -n 10
@M00176:65:000000000-A41FR:1:1101:14282:1412 2:N:0:0
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCGGAATNCCATNCCNCTCTGCNNNNNNNNNNNNNNNNNNNNNNNNN
+
#########################################################################################################<</?F/#/</?#?<#<???/<#########################
@M00176:65:000000000-A41FR:1:1101:16939:1420 2:N:0:0
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCNCNNTNCNNNNNNNNNNNGGGAATTCCACNCGCCTCTTCNNNNNNNNNNNNNNNNNNNNNNNNN
+
######################################################################################?#?##?#?###########<??/?FC>FBF#</??FAGHG#########################
@M00176:65:000000000-A41FR:1:1101:14746:1560 2:N:0:0
NNNNTTTCGTNGATNNNNNNNCNGNNNNNNNNNNNNNNNNNNNTNNNNNNNTACCNNNNNNNNTGGGCCTCTGTGCGCATACCGTTCGAGAGCGCCGGCCACGGCATTCGTGTTGCTCCTCTAAGCTGTGCGTGTTCACTGTACAACCNNN
# barcode文件内容
zcat barcodes.fastq.gz | head -n 10
@M00176:65:000000000-A41FR:1:1101:14282:1412 1:N:0:0
NNNNNNNNNNNN
+
############
@M00176:65:000000000-A41FR:1:1101:16939:1420 1:N:0:0
NNNNNNNNNNNN
+
############
@M00176:65:000000000-A41FR:1:1101:14746:1560 1:N:0:0
NNNNNNNNNNNN
数据导入:
cd emp-paired-end-sequences
qiime tools import \
--type EMPPairedEndSequences \
--input-path ./ \
--output-path emp-paired-end-sequences.qza

带barcodes的单端序列文件导入

获取样例文件
#
cd muxed-se-barcode-in-seq/
wget \
-O sequences.fastq.gz \
https://data.qiime2.org/2023.9/tutorials/importing/muxed-se-barcode-in-seq.fastq.gz
# 查看输入文件格式:
zcat sequences.fastq.gz | head -n 20
@M00899:113:000000000-A5K20:1:1101:18850:2539 1:N:0:2
GCTACGGGGGGCAGCAGTGGGGAATATTGCACAATGGGCGAAAGCCTGATGCAGCAACGCCGCGTGAACGATGAAGGTCTTCGGATCGTAAAGTTCTGTTGCAGGGGAAGATAATGACGGTACCCTGTGAGGAAGCCCCGGCTAACTACGTGCCAGCAGCCGCGGTAATACGTAGGGGGCTAGCGTTATCCGGATTTACTGGGCGTAAAGGGTGCGTAGGTGGTCCTTCAAGTCGGTGGTTAAAGGCTAAGGCTCAACCGTAGTAAGCCGCCGAAACTGGAGGACTTGAGTGAAGGAGAGG
+
-8ABCC>=>5811884:<:99=?@EECFFGDFADECFFFEEDDEFEDEDFFFEEFCCBCF>CCB3CFF:BBFFFCCD,8@9C@C:+5@@:A@C<FDCFBEG>FFFDGCCEC?FGGGGGGGGGCFGGFCFGGGGGGGGGGEG7CFFGFFFGGGFG?FACE;:8CCCCEEF9<F@FFEGGC**/:3:2CC@:C;C81;C9<?>FF8C758CGGG2:7DC>EECEFE9+27CF492/8B7>D)7@F=FFCFF*9F52<2,289<0:44AB<49(3<>F51).69D?D34*44:4<5<B?::086
@M00899:113:000000000-A5K20:1:1101:25454:3578 1:N:0:2
CCTACGGGAGGCAGCAGTGAGGAATATTGGTCAATGGGCGAGAGCCTGAACCAGCCAAGTAGCGTGCAGGATGACGGCCCTATGGGTTGTAAACTGCTTTTGTATGGGGATAAAGTCAGTCACGTGTGATTGTTTGCAGGTACCATACGAATAAGGACCGGCTAATTCCGTGCCAGCAGCCGCGGTAATACGGAAGGTCCGGGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGGCTGGAGATTAAGTGTGTTGTGAAATGTAGACGCTCAACGTCTGACTTGCAGCGCATACTGG
+
8ACCCGD@AA=18=======;CEFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGFFF?FGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCGFGGGFFFGGGGGGEFGGGGGGGGGGGGCGEGGDGGGGGGGGGG=CGGCECDGGFGGGGGGGGFGGGF>C>BFFEGFFFFF:FGGF=6=6:AFBGFFFFFFA9A<AFB?@0)>C:0<CF?C46FAD<??90;::?DA>
@M00899:113:000000000-A5K20:1:1101:25177:3605 1:N:0:2
CCTACGGGAGGCAGCAGTGAGGAATATTGGTCAATGGACGGAAGTCTGAACCAGCCAAGTAGCGTGCAGGATGACGGCCCTATGGGTTGTAAACTGCTTTTGTATGGGGATAAAGTTAGGGACGTGTCCCTATTTGCAGGTACCATACGAATAAGGACCGGCTAATTCCGTGCCAGCAGCCGCGGTAATACGGAAGGTCCAGGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGGCTGGAGATTAAGTGTGTTGTGAAATGTAGACGCTCAACGTCTGAATTGCAGCGCATACTGG
+
88BCCEDAD9018======;;CCFGGGGFGGGFGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGEGDGGGGGGGGGFFGGGGGGGGGFGGGFGGGFGGGFFGCGGGGGGFGGFDGGGGGGGGGGGGG5CBFGCGGGGC?FGGGGGGGGGGGDEGDDDGFGGGGGEGGGGGGA39>BFFDDEF4:D5@CE?CFFF>>ABGFFF9<A246<<<<B::DE=<?FGGFG4>F?DF?02211:DAF7
### barcode已经在序列中
导入文件:
qiime tools import \
--type MultiplexedSingleEndBarcodeInSequence \
--input-path sequences.fastq.gz \
--output-path multiplexed-seqs.qza

当然还有其他可导入文件格式,慢慢看吧,个人觉得有需要的时候查一下就行,不用全部了解:
qiime tools list-formats --importable
2、导出数据
官方示例,大家可以下载下来测试一下就行了,不再细讲了。

下载测试:
wget \
-O "feature-table.qza" \
"https://data.qiime2.org/2023.9/tutorials/exporting/feature-table.qza"
wget \
-O "unrooted-tree.qza" \
"https://data.qiime2.org/2023.9/tutorials/exporting/unrooted-tree.qza"
qiime tools export \
--input-path feature-table.qza \
--output-path exported-feature-table
## 导出为biom格式文件
## 参考:https://biom-format.org/documentation/format_versions/biom-2.1.html
qiime tools export \
--input-path unrooted-tree.qza \
--output-path exported-tree
## 导出为.nwk文件,可在各类软件再直接出发育树的图
head tree.nwk
(((New.CleanUp.ReferenceOTU1480:0.11995,(New.CleanUp.ReferenceOTU202:0.04479,New.CleanUp.ReferenceOTU432:0.0049)0.769:0.04661)1:0.26705,((New.CleanUp.ReferenceOTU1150:0.00016,(New.CleanUp.ReferenceOTU782:0.04264,(New.CleanUp.ReferenceOTU643:0.10438,(((New.CleanUp.ReferenceOTU1014:0.01521,New.CleanUp.ReferenceOTU270:0.02738)0.879:0.02315,(((New.CleanUp.ReferenceOTU1008:0.0378,(New.CleanUp.ReferenceOTU1222:0.01621,(New.CleanUp.ReferenceOTU230:0.01829,(New.CleanUp.ReferenceOTU1047:0.0303,New.CleanUp.ReferenceOTU605:0.00015)0.88:0.01596)0.871:0.01515)0.714:0.01214)0.996:0.06757,(New.CleanUp.ReferenceOTU681:0.00441,(New.CleanUp.ReferenceOTU1485:0.01177,New.CleanUp.ReferenceOTU130:0.05699)0.923:0.02883)0.367:0.02397)0.17:0.00665,(New.CleanUp.ReferenceOTU1330:0.0258,(New.CleanUp.ReferenceOTU903:0.04234,(((New.CleanUp.ReferenceOTU1077:0.00014,(New.CleanUp.ReferenceOTU582:0.0171,New.CleanUp.ReferenceOTU987:0.0226)0.951:0.02372)0.892:0.02206,(New.CleanUp.ReferenceOTU891:0.01038,(((New.CleanUp.ReferenceOTU1066:0.01299,New.CleanUp.ReferenceOTU764:0.02254)0.851:0.01818,(New.CleanUp.ReferenceOTU180:0.02021,New.CleanUp.ReferenceOTU535:0.00014)0.795:0.01084)0.921:0.02441,(New.CleanUp.ReferenceOTU1212:0.00521
mkdir extracted-feature-table
qiime tools extract \
--input-path feature-table.qza \
--output-path extracted-feature-table
##
3、样品元数据处理
这个可能得科学上网,给大家下载了,大家参考下载吧。
https://download.csdn.net/download/zrc_xiaoguo/88616825?spm=1001.2014.3001.5503
样例文件列表:

sample-metadata数据:

元数据表qiime2转换命令:
qiime metadata tabulate \
--m-input-file sample-metadata.tsv \
--o-visualization tabulated-sample-metadata.qzv
qiime metadata tabulate \
--m-input-file faith_pd_vector.qza \
--o-visualization tabulated-faith-pd-metadata.qzv
qiime metadata tabulate \
--m-input-file sample-metadata.tsv \
--m-input-file faith_pd_vector.qza \
--o-visualization tabulated-combined-metadata.qzv
qiime emperor plot \
--i-pcoa unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-file faith_pd_vector.qza \
--o-visualization unweighted-unifrac-emperor-with-alpha.qzv
转换后数据qzv查看



4、数据过滤
Filtering data — QIIME 2 2023.9.2 documentation
特征表过滤Filtering feature tables
基于总出现频率过滤,去掉低丰度序列,或筛选一定丰度的序列,比如说将频率低于1500的序列去掉,包括两个参数,最高和最低频率
The --p-min-frequency and --p-max-frequency can be combined to filter based on lower and upper limits of total frequency
这里只给了一个最低频率的限制。
qiime feature-table filter-samples \
--i-table table.qza \
--p-min-frequency 1500 \
--o-filtered-table sample-frequency-filtered-table.qza
基于出现的偶然性的过滤???Contingency-based filtering,也就是最少在多少个样品中都需要出现,通量有最高和最低频率设定: --p-min-features and --p-min-samples。
qiime feature-table filter-features \
--i-table table.qza \
--p-min-samples 2 \
--o-filtered-table sample-contingency-filtered-table.qza
字段筛选Identifier-based filtering, 也就是保留哪些特征数据内容。
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file samples-to-keep.tsv \
--o-filtered-table id-filtered-table.qza
同样还有很多其他的筛选方式:
Metadata-based filtering
Taxonomy-based filtering of tables and sequences
序列过滤Filtering sequences
比如说这里根据原序列文件和分类文件提取mitochondria,chloroplast这两个门水平的序列文件。
qiime taxa filter-seqs \
--i-sequences sequences.qza \
--i-taxonomy taxonomy.qza \
--p-include p__ \
--p-exclude mitochondria,chloroplast \
--o-filtered-sequences sequences-with-phyla-no-mitochondria-no-chloroplast.qza
Filtering distance matrices
qiime diversity filter-distance-matrix \
--i-distance-matrix distance-matrix.qza \
--m-metadata-file samples-to-keep.tsv \
--o-filtered-distance-matrix identifier-filtered-distance-matrix.qza
qiime diversity filter-distance-matrix \
--i-distance-matrix distance-matrix.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "[subject]='subject-2'" \
--o-filtered-distance-matrix subject-2-filtered-distance-matrix.qza
5、重要参考数据库
这个不多说了吧,大家按使用需求来下载,后面逐步会用到:
Data resources — QIIME 2 2023.9.2 documentation
6、各个插件模块使用
不熟悉的先查看help信息和官网介绍:
q2-alignment 是 QIIME 2 生态系统中的一个插件,用于对生物信息学序列数据进行比对和序列比较的工具。它可以用于将不同样本中的序列进行比对,分析它们之间的相似性和差异性。q2-alignment 提供了一些常用的比对算法和工具,使用户能够对序列进行比对、生成比对结果和后续的分析。
以下是 q2-alignment 插件的一些主要功能和使用方法:
功能和用途:
- 序列比对:对DNA、RNA或蛋白质序列进行比对。
- 多序列比对:能够处理多个序列文件,比对它们之间的相似性。
- 生成比对结果:产生比对结果,以便后续分析或可视化。
- 支持多种比对算法:包括常用的比对算法,如BLAST、MUSCLE、MAFFT 等。
qiime alignment --help
Usage: qiime alignment [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin provides support for generating and
manipulating sequence alignments.
Plugin website: https://github.com/qiime2/q2-alignment
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
mafft De novo multiple sequence alignment with MAFFT
mafft-add Add sequences to multiple sequence alignment with MAFFT.
mask Positional conservation and gap filtering.
# 运行比对(示例使用 MAFFT)
qiime alignment mafft \
--i-sequences sequences.qza \
--o-alignment aligned_sequences.qza
用于进行组成分析,尤其是在处理微生物组数据中的相对丰度数据时非常有用。该插件可以帮助用户探索和比较微生物组中微生物群落的组成。
qiime composition --help
Usage: qiime composition [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin supports methods for compositional data
analysis.
Plugin website: https://github.com/qiime2/q2-composition
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
add-pseudocount Add pseudocount to table.
ancom Apply ANCOM to identify features that differ in abundance.
ancombc Analysis of Composition of Microbiomes with Bias Correction
da-barplot Differential abundance bar plots
tabulate View tabular output from ANCOM-BC.
# 导入数据
qiime composition add-pseudocount \
--i-table table.qza \
--o-composition-table composition.qza
#数据转换
qiime composition ilr-transform \
--i-table composition.qza \
--o-transformed-table ilr_composition.qza
#可视化分析结果
qiime composition pcoa \
--i-table ilr_composition.qza \
--o-pcoa ilr_composition_pcoa.qza
qiime emperor plot \
--i-pcoa ilr_composition_pcoa.qza \
--m-metadata-file metadata.txt \
--o-visualization ilr_composition_emperor.qzv
q2-cutadapt
q2-cutadapt插件是用于对DNA序列数据进行预处理和过滤的工具。它基于Cutadapt软件,允许用户对Illumina测序数据进行裁剪(trimming)、过滤(filtering)和修剪(adapter removal)等操作,以消除低质量序列、去除适配器、修剪序列末端等。这有助于提高序列数据的质量,为后续的分析准备干净、高质量的数据。
#
qiime cutadapt --help
Usage: qiime cutadapt [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin uses cutadapt to work with adapters (e.g.
barcodes, primers) in sequence data.
Plugin website: https://github.com/qiime2/q2-cutadapt
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
demux-paired Demultiplex paired-end sequence data with barcodes in-
sequence.
demux-single Demultiplex single-end sequence data with barcodes in-
sequence.
trim-paired Find and remove adapters in demultiplexed paired-end
sequences.
trim-single Find and remove adapters in demultiplexed single-end
sequences.
qiime cutadapt trim-paired \
--i-demultiplexed-sequences demux.qza \
--p-cores 8 \
--p-front-f CCTACGGGNGGCWGCAG \
--p-front-r GACTACHVGGGTATCTAATCC \
--p-discard-untrimmed \
--o-trimmed-sequences demux_trimmed.qza
参数解释:
–i-demultiplexed-sequences: 输入的序列文件(需提前导入到QIIME 2)。
–p-cores: 并行处理的CPU核心数量。
–p-front-f 和 --p-front-r: 正向和反向引物序列。
–p-discard-untrimmed: 丢弃未被修剪的序列。
–o-trimmed-sequences: 输出修剪后的序列文件。
q2-dada2是QIIME 2的插件之一,基于DADA2算法,用于去噪和分析Illumina测序生成的16S rRNA数据。
qiime dada2 --help
Usage: qiime dada2 [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin wraps DADA2 and supports sequence quality
control for single-end and paired-end reads using the DADA2 R library.
Plugin website: http://benjjneb.github.io/dada2/
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
denoise-ccs Denoise and dereplicate single-end Pacbio CCS
denoise-paired Denoise and dereplicate paired-end sequences
denoise-pyro Denoise and dereplicate single-end pyrosequences
denoise-single Denoise and dereplicate single-end sequences
qiime dada2 denoise-single \
--i-demultiplexed-seqs input-demux.qza \
--p-trim-left 0 \
--p-trunc-len 120 \
--o-representative-sequences rep-seqs-dada2.qza \
--o-table table-dada2.qza \
--o-denoising-stats stats-dada2.qza
参数说明:
-
--i-demultiplexed-seqs:输入的demultiplexed序列文件。 -
--p-trim-left:要去除的序列的前部分碱基数量。 -
--p-trunc-len:截断序列的长度。 -
--o-representative-sequences:输出的代表序列文件。 -
--o-table:生成的特征表文件。 -
--o-denoising-stats:生成的去噪统计文件。 -
q2-deblur
q2-deblur插件通过识别和去除16S rRNA基因测序数据中的测序错误和噪声,以生成高质量的序列数据。其主要步骤包括:
- 生成特征表(Feature table): 从原始的FASTQ格式文件中导入数据,创建特征表。
- 质量过滤: 过滤低质量序列,去除低质量序列读数。
- 去噪处理: 使用Deblur算法去除测序错误和噪声,生成高质量的特征序列。
- 生成结果: 输出一个经过去噪处理和质量过滤的特征表和序列文件。
qiime deblur --help
Usage: qiime deblur [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin wraps the Deblur software for performing
sequence quality control.
Plugin website: https://github.com/biocore/deblur
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
denoise-16S Deblur sequences using a 16S positive filter.
denoise-other Deblur sequences using a user-specified positive filter.
visualize-stats Visualize Deblur stats per sample.
qiime deblur denoise-16S \
--i-demultiplexed-seqs your_qza_file.qza \
--p-trim-length 250 \ # 设置序列截断长度
--o-representative-sequences rep_seqs.qza \
--o-table table.qza \
--p-sample-stats \ # 如果需要生成样本统计信息
--o-stats deblur_stats.qza
结果数据导出
# 结果数据导出
qiime tools export --input-path rep_seqs.qza --output-path exported_rep_seqs
qiime tools export --input-path table.qza --output-path exported_table
qiime tools export --input-path deblur_stats.qza --output-path exported_stats
q2-demux 插件用于处理 DNA 或 RNA 测序数据的样本数据解复用(demultiplexing)和质量控制。这个插件允许用户根据样本的不同 DNA 或 RNA 序列标签(barcode 或者 Illumina 测序的 index)将混合测序数据集拆分成单独的样本。下面是 q2-demux 插件的基本介绍和使用步骤:
q2-demux 插件功能:
-
数据解复用(Demultiplexing):
- 将混合测序数据根据每个样本的唯一标识(barcode 或 index)分割成单独的样本序列文件。
-
质量控制:
- 提供了检查序列数据质量的功能,允许用户查看样本数据的质量分数并进行必要的处理,比如修剪或过滤低质量序列。
qiime demux --help
Usage: qiime demux [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin supports demultiplexing of single-end and
paired-end sequence reads and visualization of sequence quality information.
Plugin website: https://github.com/qiime2/q2-demux
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
emp-paired Demultiplex paired-end sequence data generated
with the EMP protocol.
emp-single Demultiplex sequence data generated with the EMP
protocol.
filter-samples Filter samples out of demultiplexed data.
partition-samples-paired Split demultiplexed sequence data into partitions.
partition-samples-single Split demultiplexed sequence data into partitions.
subsample-paired Subsample paired-end sequences without
replacement.
subsample-single Subsample single-end sequences without
replacement.
summarize Summarize counts per sample.
tabulate-read-counts Tabulate counts per sample
# 执行去重复
qiime demux emp-paired \
--i-seqs demux.qza \
--m-barcodes-file sample-metadata.tsv \
--m-barcodes-column BarcodeSequence \
--o-per-sample-sequences demux-paired-end.qza \
--o-error-correction-details demux-details.qza
## 查看结果
qiime demux summarize \
--i-data demux-paired-end.qza \
--o-visualization demux-summary.qzv
q2-diversity是其中一个用于计算和分析生物多样性的插件。它可以帮助你评估样本群落的多样性和差异性。
qiime diversity --help
Usage: qiime diversity [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin supports metrics for calculating and
exploring community alpha and beta diversity through statistics and
visualizations in the context of sample metadata.
Plugin website: https://github.com/qiime2/q2-diversity
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
adonis adonis PERMANOVA test for beta group significance
alpha Alpha diversity
alpha-correlation Alpha diversity correlation
alpha-group-significance Alpha diversity comparisons
alpha-phylogenetic Alpha diversity (phylogenetic)
alpha-rarefaction Alpha rarefaction curves
beta Beta diversity
beta-correlation Beta diversity correlation
beta-group-significance Beta diversity group significance
beta-phylogenetic Beta diversity (phylogenetic)
beta-rarefaction Beta diversity rarefaction
bioenv bioenv
core-metrics Core diversity metrics (non-phylogenetic)
core-metrics-phylogenetic Core diversity metrics (phylogenetic and non-
phylogenetic)
filter-distance-matrix Filter samples from a distance matrix.
mantel Apply the Mantel test to two distance matrices
partial-procrustes Partial Procrustes
pcoa Principal Coordinate Analysis
pcoa-biplot Principal Coordinate Analysis Biplot
procrustes-analysis Procrustes Analysis
tsne t-distributed stochastic neighbor embedding
umap Uniform Manifold Approximation and Projection
# Alpha多样性(样本内部多样性)
# 计算Shannon指数:
qiime diversity alpha \
--i-table your_feature_table.qza \
--p-metric shannon \
--o-alpha-diversity shannon_vector.qza
#Beta多样性(样本间多样性)
# 计算Bray-Curtis距离:
qiime diversity beta \
--i-table your_feature_table.qza \
--p-metric braycurtis \
--o-distance-matrix braycurtis_distance_matrix.qza
# 可视化和统计分析
qiime diversity alpha-group-significance \
--i-alpha-diversity shannon_vector.qza \
--m-metadata-file your_sample_metadata.txt \
--o-visualization shannon_group_significance.qzv
qiime diversity beta-group-significance \
--i-distance-matrix braycurtis_distance_matrix.qza \
--m-metadata-file your_sample_metadata.txt \
--o-visualization braycurtis_group_significance.qzv
q2-diversity-lib 简介
q2-diversity-lib是QIIME 2的一个插件,用于计算多样性指数和样本间的差异。它基于多种生物多样性指标来评估微生物群落的多样性,并允许用户进行统计比较和可视化。
功能特性
- 计算多样性指数:支持计算多种多样性指数,如Shannon、Simpson、Chao1等,可以帮助衡量群落内物种的多样性和丰富度。
- Beta多样性计算:计算不同样本之间的差异和相似性,例如Bray-Curtis、Jaccard、Unweighted UniFrac、Weighted UniFrac等距离指标。
- Beta多样性可视化:生成多样性分析的可视化图表,如PCoA(Principal Coordinates Analysis)图表,展示样本之间的差异。
- 组间比较:支持组间多样性比较,通过PERMANOVA(Permutational Multivariate Analysis of Variance)等方法进行样本群落的差异性分析。
qiime diversity-lib --help
Usage: qiime diversity-lib [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin computes individual metrics for community
alpha and beta diversity.
Plugin website: https://github.com/qiime2/q2-diversity-lib
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
alpha-passthrough Alpha Passthrough (non-phylogenetic)
beta-passthrough Beta Passthrough (non-phylogenetic)
beta-phylogenetic-meta-passthrough
Beta Phylogenetic Meta Passthrough
beta-phylogenetic-passthrough Beta Phylogenetic Passthrough
bray-curtis Bray-Curtis Dissimilarity
faith-pd Faith's Phylogenetic Diversity
jaccard Jaccard Distance
observed-features Observed Features
pielou-evenness Pielou's Evenness
shannon-entropy Shannon's Entropy
unweighted-unifrac Unweighted Unifrac
weighted-unifrac Weighted Unifrac
计算使用
#计算Alpha多样性(多样性指数)
qiime diversity alpha \
--i-table table.qza \
--p-metric shannon \
--o-alpha-diversity shannon_alpha.qza
#计算Beta多样性距离
qiime diversity beta \
--i-table table.qza \
--p-metric braycurtis \
--o-distance-matrix braycurtis_distance.qza
#可视化Beta多样性分析结果(例如PCoA)
qiime diversity pcoa \
--i-distance-matrix braycurtis_distance.qza \
--o-pcoa braycurtis_pcoa.qza
qiime emperor plot \
--i-pcoa braycurtis_pcoa.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization braycurtis_emperor.qzv
#进行组间多样性比较(以PERMANOVA为例)
qiime diversity beta-group-significance \
--i-distance-matrix braycurtis_distance.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization braycurtis_permanova.qzv \
--p-method permanova
qiime emperor --help
Usage: qiime emperor [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin wraps Emperor and supports interactive
visualization of ordination plots.
Plugin website: http://emperor.microbio.me
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
biplot Visualize and Interact with Principal Coordinates Analysis
Biplot
plot Visualize and Interact with Principal Coordinates Analysis
Plots
procrustes-plot Visualize and Interact with a procrustes plot
q2-feature-classifier 是 QIIME 2 中的一个插件,用于对16S rRNA或ITS等序列数据进行分类和注释。它主要用于将序列分类为特定的分类单元,比如对OTUs(操作分类单元)或者物种进行分类。
qiime feature-classifier --help
Usage: qiime feature-classifier [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin supports taxonomic classification of
features using a variety of methods, including Naive Bayes, vsearch, and
BLAST+.
Plugin website: https://github.com/qiime2/q2-feature-classifier
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
blast BLAST+ local alignment search.
classify-consensus-blast BLAST+ consensus taxonomy classifier
classify-consensus-vsearch VSEARCH-based consensus taxonomy classifier
classify-hybrid-vsearch-sklearn
ALPHA Hybrid classifier: VSEARCH exact match
+ sklearn classifier
classify-sklearn Pre-fitted sklearn-based taxonomy classifier
extract-reads Extract reads from reference sequences.
find-consensus-annotation Find consensus among multiple annotations.
fit-classifier-naive-bayes Train the naive_bayes classifier
fit-classifier-sklearn Train an almost arbitrary scikit-learn
classifier
makeblastdb Make BLAST database.
vsearch-global VSEARCH global alignment search
#训练一个 SILVA 数据库上的分类器可以使用如下命令
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads silva-132-99-515-806-nb-classifier.qza \
--i-reference-taxonomy silva-132-99-515-806-nb-classifier.qza \
--o-classifier classifier.qza
# 使用训练好的分类器对样本数据进行分类
qiime feature-classifier classify-sklearn \
--i-classifier classifier.qza \
--i-reads paired-end-demux.qza \
--o-classification taxonomy.qza
q2-feature-table 插件是 QIIME 2 中的一个重要插件,用于处理和操作特征表(feature table)数据,它包括了许多功能,用于对微生物组数据进行分析和可视化。
以下是 q2-feature-table 插件的一些主要功能和使用方法:
- 导入特征表数据: 可以使用该插件将不同格式的特征表数据导入到 QIIME 2 中,如BIOM格式、文本格式、或其他常见格式的特征表。
- 特征表汇总和统计: 可以对特征表进行汇总和统计描述,比如计算每个样本中的特征数量、每个特征在样本中的出现频率等。
- 特征表的过滤和修剪: 提供了多种方法对特征表进行过滤和修剪,如去除低频特征、去除低丰度特征、保留指定样本数或特征数等。
- 特征表的转换和变换: 可以对特征表进行转换,如转置、归一化、对数转换等,以适应不同类型的分析需求。
- 特征表的合并和拆分: 可以将多个特征表合并为一个,也可以根据样本元数据信息将特征表拆分为多个子集。
- 特征表的可视化: 支持对特征表进行可视化展示,比如生成特征数量分布图、绘制热图展示特征在样本中的丰度等。
qiime feature-table --help
Usage: qiime feature-table [OPTIONS] COMMAND [ARGS]...
Description: This is a QIIME 2 plugin supporting operations on sample by
feature tables, such as filtering, merging, and transforming tables.
Plugin website: https://github.com/qiime2/q2-feature-table
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
core-features Identify core features in table
filter-features Filter features from table
filter-features-conditionally Filter features from a table based on
abundance and prevalence
filter-samples Filter samples from table
filter-seqs Filter features from sequences
group Group samples or features by a metadata
column
heatmap Generate a heatmap representation of a
feature table
merge Combine multiple tables
merge-seqs Combine collections of feature sequences
merge-taxa Combine collections of feature taxonomies
presence-absence Convert to presence/absence
rarefy Rarefy table
relative-frequency Convert to relative frequencies
rename-ids Renames sample or feature ids in a table
split Split one feature table into many
subsample Subsample table
summarize Summarize table
tabulate-seqs View sequence associated with each feature
transpose Transpose a feature table.
#特征表的摘要和统计信息
qiime feature-table summarize \
--i-table feature-table.qza \
--o-visualization feature-table-summary.qzv
#特征表的过滤和修剪
qiime feature-table filter-features \
--i-table feature-table.qza \
--p-min-frequency 10 \
--o-filtered-table filtered-feature-table.qza
qiime feature-table filter-samples \
--i-table feature-table.qza \
--p-min-frequency 500 \
--o-filtered-table filtered-sample-table.qza
# 特征表的合并和操作
qiime feature-table merge \
--i-tables table1.qza \
--i-tables table2.qza \
--o-merged-table merged-table.qza
#计算特征表的β-diversity:
qiime diversity beta \
--i-table feature-table.qza \
--o-distance-matrix beta-diversity.qza \
--p-metric braycurtis
#可视化特征表
qiime feature-table summarize \
--i-table feature-table.qza \
--o-visualization feature-table-summary.qzv
#可视化β-diversity距离矩阵
qiime diversity beta-phylogenetic \
--i-table feature-table.qza \
--i-phylogeny rooted-tree.qza \
--o-distance-matrix beta-diversity.qza \
--p-metric unweighted_unifrac
qiime diversity pcoa \
--i-distance-matrix beta-diversity.qza \
--o-pcoa pcoa-results.qza
qiime emperor plot \
--i-pcoa pcoa-results.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization emperor.qzv
q2-fragment-insertion插件是用于将未分配的DNA序列(通常是16S rRNA或18S rRNA序列)嵌入(插入)到预先构建的参考进化树中的工具。这个插件可以帮助解决一些问题,比如通过将未知序列嵌入到进化树中,来推断未知序列的系统发育位置。
qiime fragment-insertion --help
Usage: qiime fragment-insertion [OPTIONS] COMMAND [ARGS]...
Description: No description available. See plugin website:
https://github.com/qiime2/q2-fragment-insertion
Plugin website: https://github.com/qiime2/q2-fragment-insertion
Getting user support: https://github.com/qiime2/q2-fragment-insertion/issues
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
classify-otus-experimental Experimental: Obtain taxonomic lineages, by
finding closest OTU in reference phylogeny.
filter-features Filter fragments in tree from table.
sepp Insert fragment sequences using SEPP into
reference phylogenies.
qiime fragment-insertion sepp \
--i-representative-sequences <代表序列文件.qza> \
--i-reference-database <参考数据库.qza> \
--o-tree <输出进化树.qza> \
--o-placements <输出插入位置文件.qza>
-
--i-representative-sequences是代表性序列文件的位置。 -
--i-reference-database是参考数据库文件的位置。 -
--o-tree指定输出的进化树文件。 -
--o-placements指定输出的插入位置文件。 -
q2-longitudinal
q2-longitudinal 插件是 QIIME 2 中的一个插件,专门用于处理微生物组长期研究的数据。该插件允许用户对时间序列实验数据进行分析,以便检测微生物组随时间变化的情况,比较不同条件下的变化,以及对这些变化的统计显著性进行评估。
以下是 q2-longitudinal 插件的一些主要功能和使用命令:
主要功能:
- 时间序列数据可视化:生成时间序列样本数据的可视化图表,比如长期研究的变化趋势、样本之间的差异等。
- 差异分析:比较不同时间点或不同处理组之间的微生物组成差异。
- Alpha 和 Beta 多样性分析:评估微生物群落在时间序列中的多样性和相似性变化。
- 线性混合效应模型:对微生物组数据进行线性模型分析,以研究时间、处理效应和其交互作用对微生物组成的影响。
qiime longitudinal --help
Usage: qiime longitudinal [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin supports methods for analysis of time
series data, involving either paired sample comparisons or longitudinal
study designs.
Plugin website: https://github.com/qiime2/q2-longitudinal
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
anova ANOVA test
feature-volatility Feature volatility analysis
first-differences Compute first differences or difference from
baseline between sequential states
first-distances Compute first distances or distance from baseline
between sequential states
linear-mixed-effects Linear mixed effects modeling
maturity-index Microbial maturity index prediction.
**先自我介绍一下,小编浙江大学毕业,去过华为、字节跳动等大厂,目前在阿里**
**深知大多数程序员,想要提升技能,往往是自己摸索成长,但自己不成体系的自学效果低效又漫长,而且极易碰到天花板技术停滞不前!**
**因此收集整理了一份《2024年最新Linux运维全套学习资料》,初衷也很简单,就是希望能够帮助到想自学提升又不知道该从何学起的朋友。**





**既有适合小白学习的零基础资料,也有适合3年以上经验的小伙伴深入学习提升的进阶课程,涵盖了95%以上运维知识点,真正体系化!**
**由于文件比较多,这里只是将部分目录截图出来,全套包含大厂面经、学习笔记、源码讲义、实战项目、大纲路线、讲解视频,并且后续会持续更新**
**[需要这份系统化的资料的朋友,可以点击这里获取!](https://bbs.csdn.net/topics/618542503)**
and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
anova ANOVA test
feature-volatility Feature volatility analysis
first-differences Compute first differences or difference from
baseline between sequential states
first-distances Compute first distances or distance from baseline
between sequential states
linear-mixed-effects Linear mixed effects modeling
maturity-index Microbial maturity index prediction.
**先自我介绍一下,小编浙江大学毕业,去过华为、字节跳动等大厂,目前在阿里**
**深知大多数程序员,想要提升技能,往往是自己摸索成长,但自己不成体系的自学效果低效又漫长,而且极易碰到天花板技术停滞不前!**
**因此收集整理了一份《2024年最新Linux运维全套学习资料》,初衷也很简单,就是希望能够帮助到想自学提升又不知道该从何学起的朋友。**
[外链图片转存中...(img-TDKg6wH1-1715144352253)]
[外链图片转存中...(img-vm8MgaIU-1715144352254)]
[外链图片转存中...(img-MVJRGhcl-1715144352255)]
[外链图片转存中...(img-Ps7GQUPK-1715144352255)]
[外链图片转存中...(img-iGkVxhaC-1715144352256)]
**既有适合小白学习的零基础资料,也有适合3年以上经验的小伙伴深入学习提升的进阶课程,涵盖了95%以上运维知识点,真正体系化!**
**由于文件比较多,这里只是将部分目录截图出来,全套包含大厂面经、学习笔记、源码讲义、实战项目、大纲路线、讲解视频,并且后续会持续更新**
**[需要这份系统化的资料的朋友,可以点击这里获取!](https://bbs.csdn.net/topics/618542503)**















 1175
1175

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









