






可以这么说,目前在长度长的三代测序领域内,基本上是PacBio和Nanopore的天下。二者测序技术各有千秋,我就来谈一点浅薄的认识。
首先,介绍一下二者的测序原理。
PacBio采用SMRT测序技术,名称取自single molecule real time sequencing,即单分子实时测序。
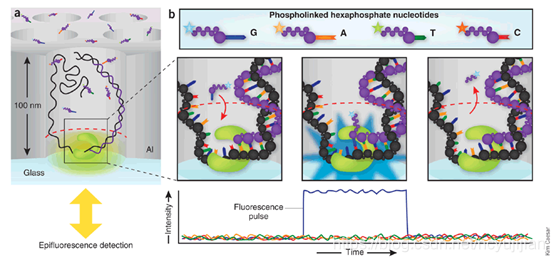
上图为PacBio的核心测序原理,基本上包括以下几个要点:
- 制造出一个ZMW孔(Zero-Mode Waveguides),一个光学物理概念。大白话说,就是在这个孔底部照射激光,这些光不会穿透小孔。这样每个孔不同颜色的光互相不会干扰。
- 四种不同的碱基,经过修饰荧光基团,在ZMW孔内,进行聚合反应的时候,荧光基团脱落。在激光的照射下,显示不同的颜色。从而可以根据颜色的变化顺序,推导DNA序列。
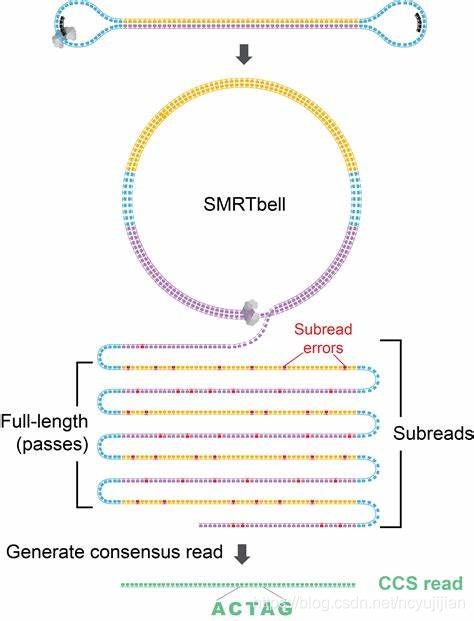
- 构建环装文库。任何高通量测序,都必须告诉机器从哪里开始测序。这个就要靠文库构建来解决。PacBio的文库结构如下图所示:
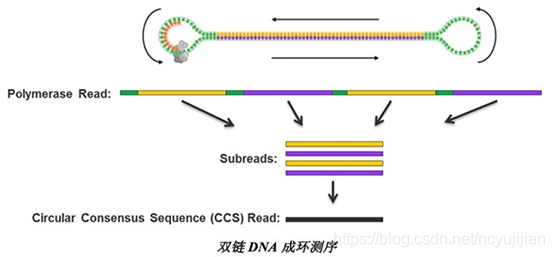
接下来,我介绍一下,nanopore的测序原理。
Nanopore也是有一个很小的纳米孔,不过这个孔略有区别。如下图所示:
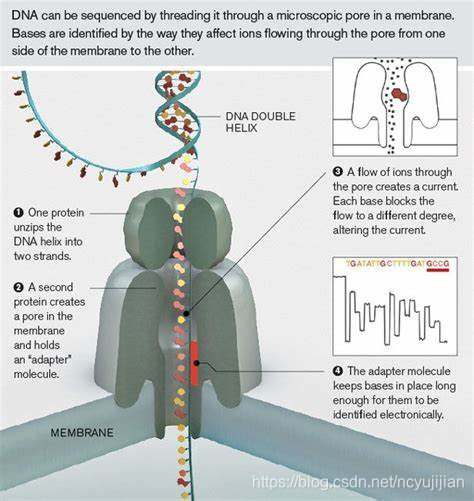
这个孔是一个天然的蛋白,孔径很小,仅能通过单链DNA分子。其测序原理通俗解释,就是想办法让单链DNA通过这个纳米孔。但是,纳米孔两侧有一个记录电压变化的传感器。在施加一个电压的时候,单链DNA的不同碱基通过纳米孔的时候,电压的变化情况是不一样的。传感器就是记录这些不一样的电压变化,从而将电信号转化为碱基信息。
同样,为了测序,nanopore测序仪也是要建库的,其文库构建过程如下图:
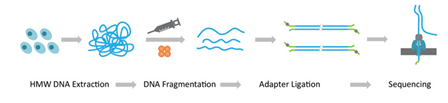
备注:Nanopore文库两侧是添加了Y字形接头的,不同于Illumina的Y字形文库,该文库携带了一个马达蛋白(具有解旋功能,将双链DNA解开变成单链,并通过纳米孔)。
介绍完了两个平台的基本原理,我来聊聊两个平台的技术特点吧。
-
PacBio可以引入CCS测序模式,即不断的滚环测序,对一个DNA分子的正反链不断测序,可以提高但碱基精度。
-
NanoPore可以做的比较小巧,且由于其测序不需要进行合成反应,不需要添加一些修饰的碱基,所以使用相对方便。不需要花费太多经费采购仪器。且由于测序仪器可以很小巧,甚至可以在野外进行测序。
-
至于读长和测序精度,实测数据来看,应该不分伯仲吧。
-
两台机器都可以直接读取甲基化信息。
-
但是,两台机器对于连续相同的碱基,读取容易出错。

















 9976
9976

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









