








目录
摘要
目前,分析由许多细胞和样品组成的单细胞数据,以及解决批次效应和不同样品制备引起的变化是一个挑战。为此,论文提出了SAUCIE,这是一种深度神经网络,它结合了神经网络提供的并行性和可伸缩性,以及数据的深度表示,这些数据可以通过学习来执行许多单细胞数据分析任务。正则化(惩罚)使得在神经网络的隐藏层中学习到的特征可以解释。在大型多患者数据集上,SAUCIE的各种隐藏层包含去噪和批量校正数据、低维可视化和无监督聚类,以及可用于探索数据的其他信息。我们分析了来自印度登革热患者的1100万个T细胞组成的180个样本数据集,并用质谱仪进行了测量。SAUCIE可以批量纠正和识别急性登革热感染的集群特征,并创建一个患者簇,对登革热的免疫反应进行分层。
引言
处理高维和大规模的单细胞数据本身就很困难,尤其是考虑到数据中的噪声、批量效应、稀疏性和异质性的程度。此外,当人们试图在样本之间进行比较时,这种影响会加剧,因为样本本身包含细胞群中嘈杂的异质成分。深度学习作为一种处理现代生物数据集的大小和维度的技术提供了希望。然而,深度学习在无监督的探索性任务中没有得到充分利用。
论文使用了一个正则化的自动编码器,这是一个神经网络,它通过一个低维瓶颈层重新创建自己的输入,该瓶颈层学习数据的表示,并实现输入的去噪重建。
由于自编码器可以无监督学习特征,揭示数据中的结构,而无需像其他降维方法那样在原始数据空间中定义或明确学习相似性或距离度量(例如,PCA使用协方差,扩散映射diffusion map基于核选择)。使用自动编码器方法构建SAUCIE,这是一种用于无监督聚类clustering、插补imputation和嵌入embedding的稀疏自动编码器,旨在通过其设计实现探索性任务。SAUCIE是一个多任务的神经网络,其输入层是单个细胞的单细胞测量数据,如质谱仪或单细胞RNA测序。不同的层显示不同的数据表示:用于可视化、批量校正、聚类和去噪。SAUCIE提供了数据的统一表示,在不同层中强调不同的方面或特征,形成一个一步式的数据分析pipeline。
论文将SAUCIE应用于登革热黄病毒研究中的1100万细胞质量流式细胞仪数据集的批量校正、去噪和聚类,该数据集包含来自40名受试者的180个样本集。
结果
SAUCIE的架构和layer的正则
为了以可伸缩的方式实现无监督学习,论文的方法基于自动编码器实现。一个关键的挑战是从模型的内部数据表示中提取有意义的表示。具体来说,我们在隐藏层中寻找表示,这些表示对于执行与单细胞数据相关的各种分析任务非常有用。在这里,自动编码器架构(图1)介绍了几个设计决策和新的正则化,以约束四个关键任务的学习表示:聚类、批量校正、去噪和插补,以及可视化和降维。对于每项任务,都会使用专门的设计决策来产生理想的结果。
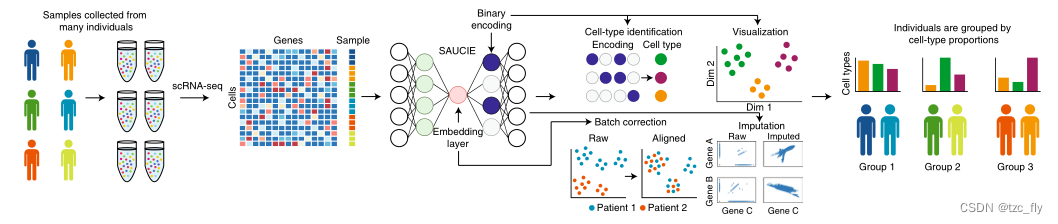
在上图中,SAUCIE使用统一的模型对整个患者队列进行插补和去噪、批量效应消除、聚类和可视化,并能够为每个受试者或受试者组提供可解释、可量化的指标。
聚类Clustering
首先,为了对数据进行聚类,引入了信息维(ID,information dimension)正则化,这鼓励了网络隐藏层中神经元的激活可以被二值化。通过获得“数字离散”二进制编码,我们可以很容易地将这些编码转换成集群。没有正则化的网络倾向于以分布式或“模拟连续”方式存储其信息。通过ID正则化,激活都接近0或1,即二进制或数字digital,因此可以通过简单的基于阈值的二值化进行聚类(图2)。
- 图a表示:应用于稀疏编码层的ID正则化产生用于聚类的数字代码;
- 图b表示:信息瓶颈,即较小的嵌入层,使用降维在输出端生成去噪数据;
- 图c表示:MMD正则化可移除批处理瑕疵。
- 图d表示:应用于去噪数据的簇内正则化提供了更清晰的簇。

这可以有效地表示数据空间的细胞的聚类(图3):

对于上图,论文将SAUCIE执行的不同分析任务与其他方法进行比较: - 图a:对Shekhar(上图)和Zeisel(下图)的数据进行聚类性能比较,样本量分别为27499和3005;
- 图b:SAUCIE与各种可视化方法在可视化性能上的比较,数据集与图a相同;
- 图c:在插补方法(补全缺失的数据)上的比较;
因此,ID正则化实现了模拟到数字的转换,从而能够将表示解释为对应于每个二进制编码的簇。为了进一步鼓励簇成为同质的细胞组,我们还引入了簇内距离正则化,该正则化将惩罚在原始数据空间中具有相似聚类层表示的细胞。
Batch correction
批次效应通常是在不同实验运行下测量的生物数据中发现的系统性差异,主要是由于环境条件,如温度、机器校准或测量效率的日常变化。因此,即使是来自非常相似的系统(例如来自同一患者的血细胞)的测量结果,在两次不同的实验运行之间似乎也存在变化或差异。为了解决这个问题,论文引入了一个最大平均差异(MMD)校正,它惩罚样本内部激活概率分布之间的差异。之前的工作尝试通过最小化MMD进行批量校正。但过去的工作都假设批次效应是微小的。相比之下,SAUCIE中额外的自动编码器重建惩罚迫使模型在每个批次中保留原始结构,一方面平衡使两个批次相似的目标,另一方面不改变它们。我们注意到,生物批次(测量或运行的数据)的概念不同于随机梯度下降中用于训练神经网络的小批次。这里,术语中的批次batch专门用于描述生物批次,当使用随机梯度下降进行训练时,使用术语中的小批次mini-batch。
在批次校正之前分析数据可能会导致误导性结果,因为批次效应的人为变化可能会淹没生物学中相关的信息变化(图4)。直接在输入空间上惩罚MMD是解决批量效应的一种有缺陷的方法,因为它需要在输入点上做出有意义的距离和相似性度量的假设。由于数据是有噪声的,而且可能是稀疏的,通过在网络的内部层上惩罚MMD,我们可以通过在这些层中表示的数据流形上对齐点来纠正复杂的、高度非线性的批处理效应。

图4展示了SAUCIE的批次修正的能力:
- 图a:SAUCIE batch correction将完美重建perfect reconstruction(这将使批次不被校正)与完美混合perfect blending(这将消除数据中的所有原始结构)相平衡,以消除技术上的批次效应,同时保留生物差异。
- 图b:增加 λ b \lambda_{b} λb(MMD正则化的幅度)对41721号登革热数据的影响。
- 图c:通过指定方法对合成GMM数据(大小为2000,上)和登革热数据(下)进行批量校正的结果。
Imputation and denoising
接下来,论文利用了一个事实,即自编码器不能准确地重建其输入,而是必须学习数据的低维表示,并对该表示进行解码以进行数据重建。这意味着重构是输入的去噪版本,因此自然可以解决影响许多真实数据(尤其是单细胞RNA测序数据)的丢失和其他噪声问题。图3中绘制的基因-基因关系(Gene-Gene)表明,尽管数据中存在噪音,但SAUCIE能够恢复基因之间有意义的关系。因此,一些下游活动,如差异基因表达,现在由这些改进的表达谱增强。
可视化
最后,论文将自动编码器的信息瓶颈层设计为二维,使其成为数据的可视化和非线性嵌入。因为网络必须根据这种内部表示准确地重建输入,所以它必须将有关细胞的所有信息压缩到这两个维度,而不像PCA或扩散图Diffusion Maps这样的方法,后者明确地保留一些未建模的变化。因此,存储的信息也是全局性的,这意味着SAUCIE可视化中靠得很近的点比相距较远的点更相似,而在tSNE等局部方法中,这在局部区域之外是不正确的。使用SAUCIE灵活学习并准确反映数据结构的能力如图3所示。
对感染登革热的患者的免疫反应分析
论文用一个大型数据集来说明SAUCIE的可扩展性,该数据集由来自45名受试者的1100万T细胞的单细胞因子测量组成,其中包括一组急性感染登革热病毒的受试者和来自同一流行地区的健康对照者。这种登革热数据是一种理想的SAUCIE测试案例,因为样本是在几个月内收集的,在不同的日期进行测量,并从印度运输。因此,迫切需要对患者分层进行批量校正和数据清理,以及统一处理、聚类和荟萃分析。
受试者之间的不同聚类比例
论文首先使用SAUCIE的MMD正则化对数据进行校正和去噪,获得各组的聚类特征,然后进一步分析它们作为单细胞版本的血液生物标记物的标记物富集。为了对细胞进行聚类,之前引入了两种新的正则化:ID正则化和within-cluster距离正则化(簇内距离正则)。这些都与自动编码器的重建惩罚相平衡,系数控制损失的每个部分的权重。对于ID正则化和簇内距离正则化,我们分别将这些系数称为 λ c \lambda_{c} λc和 λ d \lambda_{d} λd。这两种正则化会影响结果的簇数。对于给定的 λ d \lambda_{d} λd值,随着 λ c \lambda_{c} λc的增加,簇的数量减少(粗粒度)。 λ d \lambda_{d} λd的值越高,产生的团簇越多(粒度越细)。这两者一起充当旋钮,可以对其进行调整,以获得所需的簇粒度。对于这里考虑的聚类,我们使用 λ c \lambda_{c} λc为0.1, λ d \lambda_{d} λd为0.2的粗粒度聚类。如果需要其他粒度,可以使用其他系数。
论文寻找在急性期和恢复期的T细胞簇。这些可能是在感染或恢复过程中起重要作用的细胞群。论文检查每个聚类的标记丰度分布,以确定亚群。
论文在T细胞群中发现了20个集群,其中5个是CD8 T细胞,13个是CD4 T细胞。此外,有六个簇是CD4 − CD8− T细胞,其中四个是相对罕见的γδ T细胞。这些被认为是对病毒感染的反应的一个特征。有12个集群代表效应记忆细胞和9个调节性T细胞,它们是CD4+Foxp3+。其中两个簇是naive T细胞。其中几个群体表明急性、恢复期和健康受试者之间存在差异,并可用于表征这些群体中每个人的反应性质。例如,一组在早期免疫反应中起重要作用的重要但罕见的细胞,γδT细胞,被鉴定并聚集。
我们还可以可视化患者气管上的细胞水平簇比例(图5)。在这里,我们看到排列在这个流形上的簇比例揭示了在空间中不断变化的簇。该分析清楚地表明,第1簇代表急性受试者,第5簇代表健康受试者。此外,我们可以在急性感染后测量同一个体,然后在恢复期时间点进行评估(图5)。从这个角度来看,我们发现大多数受试者在急性感染时比在恢复期时更容易出现第11簇。
图5中,在11,228,838个细胞的整个登革热数据集上。
- 第一行,通过SAUCIE聚类比例确定的患者流形,通过PCA可视化,从左到右是:急性、健康、恢复期,所有受试者的合并。
- 中间一行,相同的患者集合显示为每个患者的簇比例。
- 下一行,有匹配样本的患者的急性期(x轴)与恢复期(y轴)聚类比例的比较。

可视化
SAUCIE可以处理所有受试者的细胞,构建细胞流形并提取其特征。首先,我们使用二维可视化层来可视化这个流形。(图6)分为两个embedding图,分别显示了急性和健康受试者的细胞流形。可以看出,当并排比较embedding时,有一个明显的特征变化。急性受试者缺少健康受试者体内的细胞群,反之亦然。

- 图a:通过二维SAUCIE embedding层识别急性、健康和恢复期受试者T淋巴细胞亚群的细胞流形。
- 图b:显示沿水平轴的簇和沿垂直轴的标记gene的热图。簇大小表示为热图下方的颜色栏。
- 图c:急性、恢复期和健康患者的聚类比例 。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









