







一、TCGA数据分析流程,从数据下载到差异可视化!-- UCSC xena
TCGA: 癌症基因组图谱
癌症基因组图谱 (TCGA) 是一项具有里程碑意义的癌症基因组学计划,它对 20,000 多个原发性癌症和 33 种癌症类型的正常样本进行了分子表征。NCI 和美国国家人类基因组研究所的这项合作始于 2006 年,汇集了来自不同学科和多个机构的研究人员。
在接下来的十几年里,TCGA 生成了超过 2.5PB 的基因组、表观基因组、转录组和蛋白质组数据。这些数据已经提高了我们诊断、治疗和预防癌症的能力,并将一直向公众开放,供研究界的任何人使用。
其中TCGA选择研究的33种癌症类型如下:
本期我们主要介绍如何通过UCSC xena对TCGA数据进行下载到差异分析可视化!后续我们会更新如何使用R语言的TCGAbiolinks包下载和TCGA官方下载工具gdc-client进行下载!
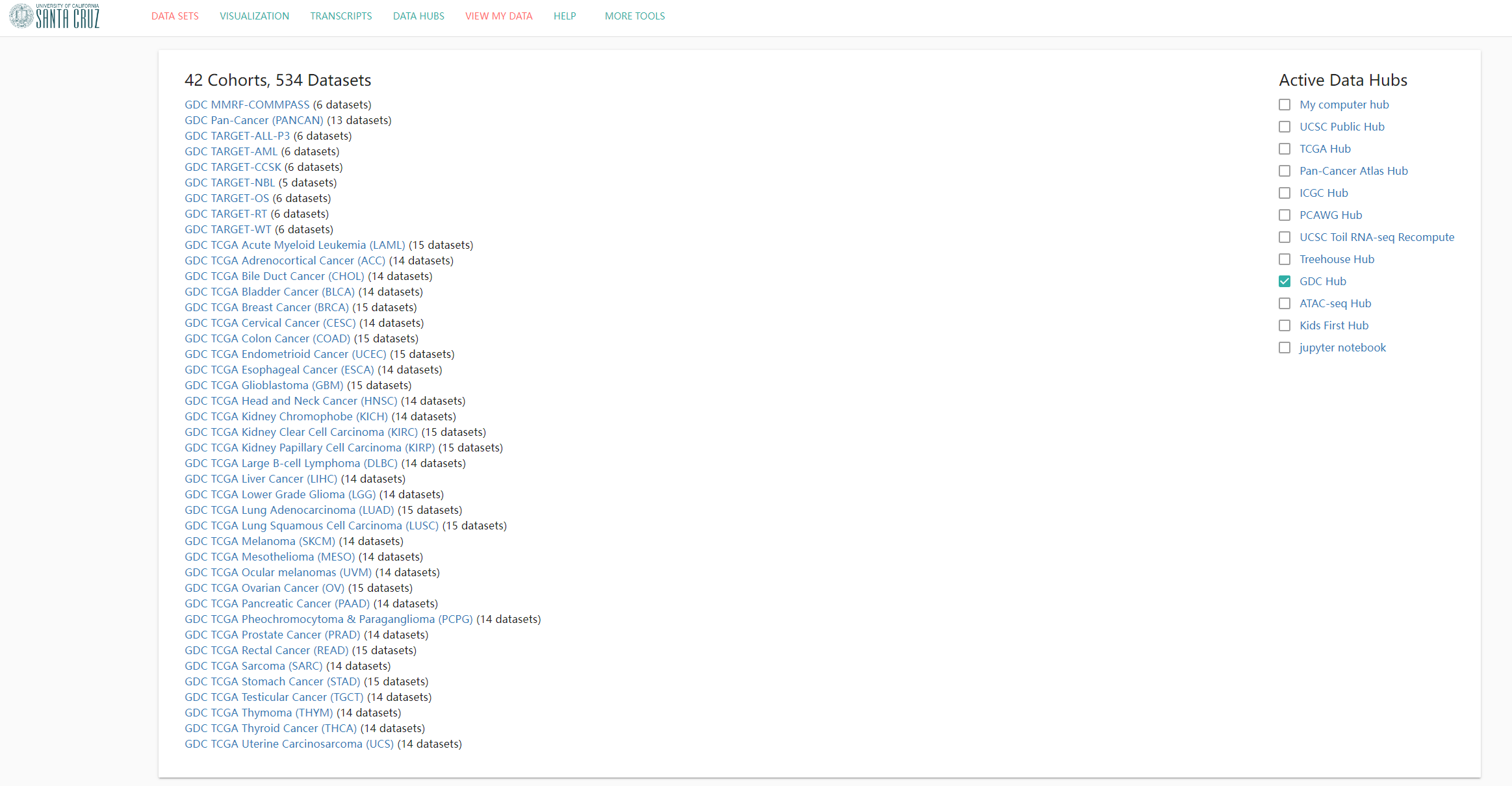
1、对于新手友好最好使用UCSC xena进行下载,其已经整理好了TCGA的数据,不需要用户自行整理,节约时间,但数据并不是最新数据!
2、对于具有R语言编程能力的用户,TCGAbiolinks能解决gdc-client下载卡顿不成功的问题,并且提供merge功能,方便后续处理,首推这个方法!
3、TCGA的官方下载工具gdc-client,使用此工具下载到的数据是最新数据,但流程繁琐,并具有较好的网络环境,需要自己单个文件merge!
UCSC xena
首先我们进入到UCSC xenahttps://xenabrowser.net/datapages/的数据集页面,推荐选择GDC-hub的数据集。
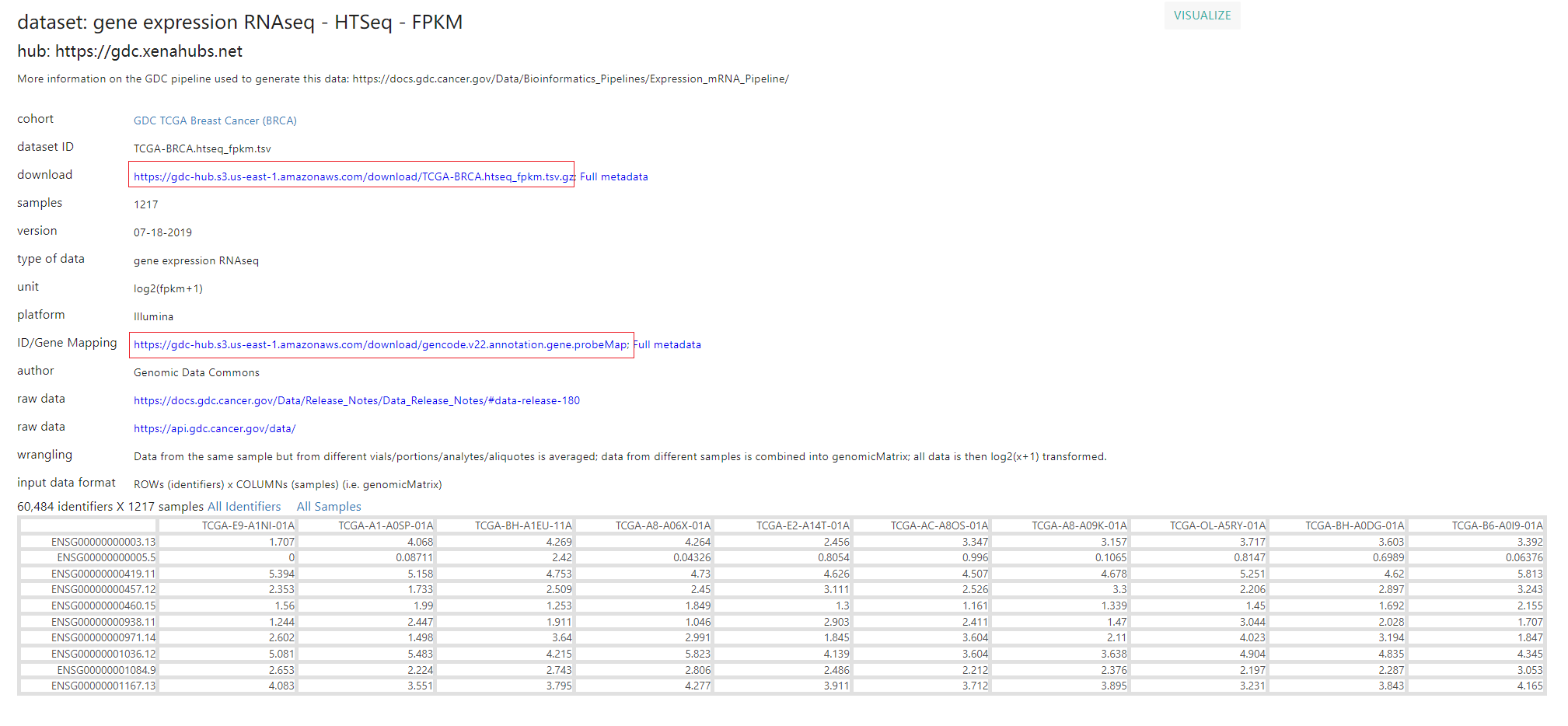
对于GDC-hub数据,UCSC对如何处理的数据进行了描述,此处我们以BRCA乳腺癌的FPKM为例,其主要使用了log2(fpkm+1)处理,方便直接进行后续的分析!

对于数据部分就不做更多的介绍了,需要更多的了解可以到其网站进行相应的了解。接下来我们开始正式的流程!
mRNA表达谱的下载
我们主要从mRNA数据详情页面对表达数据的FPKM进行下载,我们同时也下载了基因注释文件,方便ID转换。当然也可以下载count数据,对于不同的表达数据可以根据自己需求进行下载。
数据处理
1、数据导入
rm(list = ls());gc()
options(stringsAsFactors = F)
library(data.table)
library(tidyverse)
# 读取Xena下载的FPKM表达谱数据
TCGA_BRCA <- fread("TCGA-BRCA.htseq_fpkm.tsv.gz") %>% as.data.frame()
dim(TCGA_BRCA)
TCGA_BRCA[1:6, 1:4]
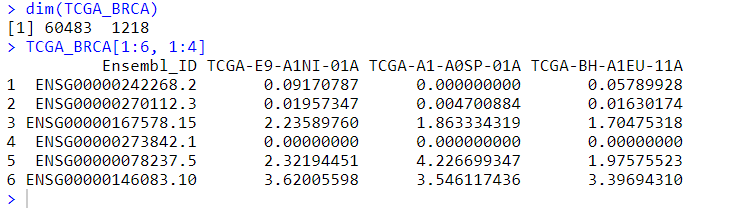
可以查看数据大小范围,标准化后的数据一般是0~20之间
2、ID转换
TCGA使用的事Ensembl_ID,为了后续分析方便,我们将其转换为gene symbol
# ID转换
probeMap <- read.delim("gencode.v22.annotation.gene.probeMap")
TCGA_BRCA <- TCGA_BRCA %>%
inner_join(probeMap, by = c("Ensembl_ID" = "id")) %>%
select(gene, starts_with("TCGA"))
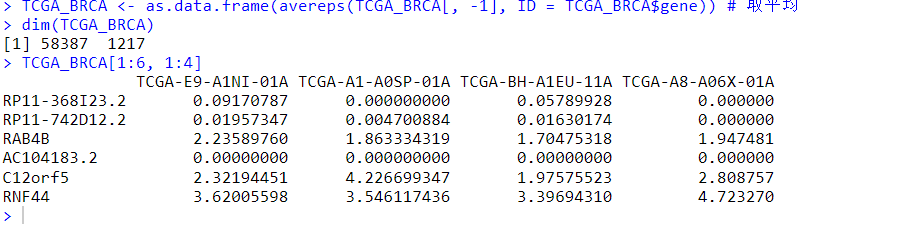
由于可能存在多个Ensembl_ID对应同一个gene symbol,因此我们对重复基因取平均值进行代替
library(limma)
TCGA_BRCA <- as.data.frame(avereps(TCGA_BRCA[, -1], ID = TCGA_BRCA$gene)) # 取平均
dim(TCGA_BRCA)
TCGA_BRCA[1:6, 1:4]
# 对于重复基因的处理还可以使用其他处理方式,如取表达值最大的一个
# 可以看到去除重复之后还剩58387个基因
3、分组信息的构建
对于TCGA样本的命名规则如下所示

·01:Sample, 这两个数字可以说是最关键、最被大家注意的,其中编号0109表示肿瘤,1019表示正常对照。
在TCGA样本中,最常见的是01和11
TCGA_group_list <- ifelse(as.numeric(substring(colnames(TCGA_BRCA),14,15)) < 10,
"Tumor","Normal") %>%
factor(.,levels = c("Normal","Tumor"))
table(TCGA_group_list)

4、limma包进行差异分析
library(limma)
group <- TCGA_group_list
design <- model.matrix(~0+group)
rownames(design)<-colnames(TCGA_BRCA)
colnames(design) <- levels(group)
dgelist <- DGEList(counts = TCGA_BRCA, group = group) %>% calcNormFactors()
fit <- voom(dgelist, design, plot = TRUE, normalize = "quantile") %>% lmFit(design)
constrasts <- paste(rev(levels(group)), collapse = "-")
cont.matrix <- makeContrasts(contrasts = constrasts, levels = design)
fit2 <- contrasts.fit(fit, cont.matrix) %>% eBayes()
DEG = topTable(fit2, coef=constrasts, n=Inf) %>% na.omit()
DEG<-DEG[(DEG$P.Value < 0.05),]
DEG$Genes <- rownames(DEG)
DEG$regulate <- ifelse(DEG$P.Value > 0.05, "unchanged",
ifelse(DEG$logFC > 1, "up-regulated",
ifelse(DEG$logFC < -1, "down-regulated", "unchanged")))
head(DEG)

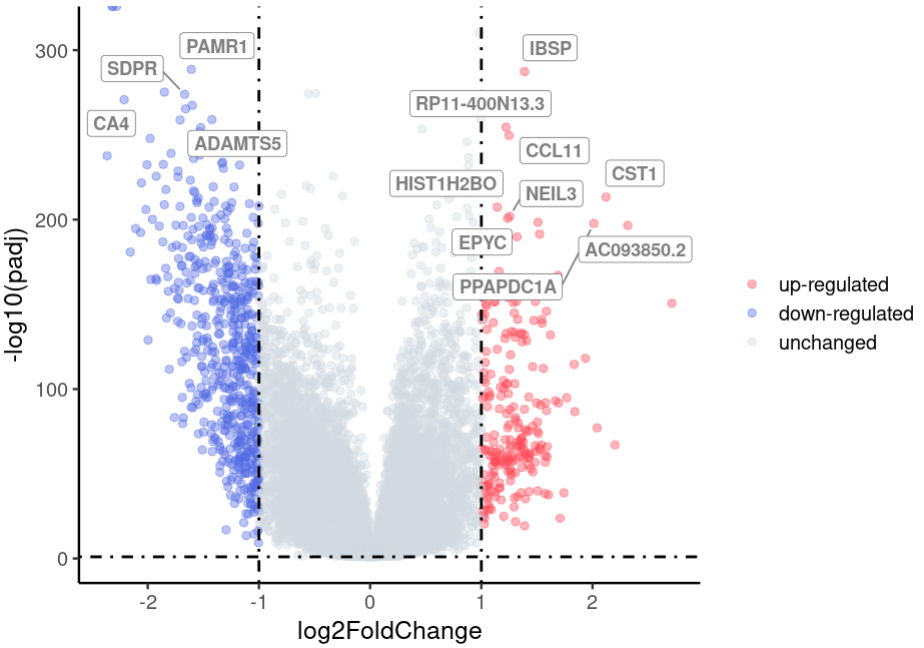
5、差异基因可视化–火山图和热图
# 火山图
data<-DEG
data$sign <-NA
data <- data[order(data$P.Value, data$logFC, decreasing = c(FALSE, TRUE)), ]
# 筛选logFC上调的基因(logFC > 1),并按p值降序排列
upregulated_genes <- data[data$logFC > 1, ]
upregulated_genes <- upregulated_genes[order(upregulated_genes$P.Value, decreasing = FALSE), ]
# 选择p值最大的前10个上调基因
top10_upregulated <- head(upregulated_genes, 10)
# 筛选logFC下调的基因(logFC < -1),并按p值降序排列
downregulated_genes <- data[data$logFC < -1, ]
downregulated_genes <- downregulated_genes[order(downregulated_genes$P.Value, decreasing = FALSE), ]
# 选择p值最大的前10个下调基因
top10_downregulated <- head(downregulated_genes, 10)
# 输出前10个上调和前10个下调基因
top10_upregulated
top10_downregulated
data$sign[which(rownames(data)%in% c(top10_upregulated$Genes, top10_downregulated$Genes) )]<-rownames(data)[which(rownames(data)%in% c(top10_upregulated$Genes, top10_downregulated$Genes) )]
library(ggrepel)
ggplot(data = data, aes(x = logFC, y = -log10(P.Value), color = regulate)) +
geom_point(alpha=0.4, size = 2) +
geom_hline(yintercept = 1,lty=4,col="black",lwd=0.8) +
geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +
theme_classic(base_size = 15) +
theme(panel.grid.minor = element_blank(),panel.grid.major = element_blank()) +
labs(x="log2FoldChange",y="-log10(padj)")+
scale_color_manual(name = "", values=c( "#ff4757","#546de5","#d2dae2"), limits = c("up-regulated", "down-regulated", "unchanged")) +
geom_label_repel(aes(label=sign), fontface="bold", color="grey50", box.padding=unit(0.35, "lines"), point.padding=unit(0.5, "lines"), segment.colour = "grey50")
# 差异热图
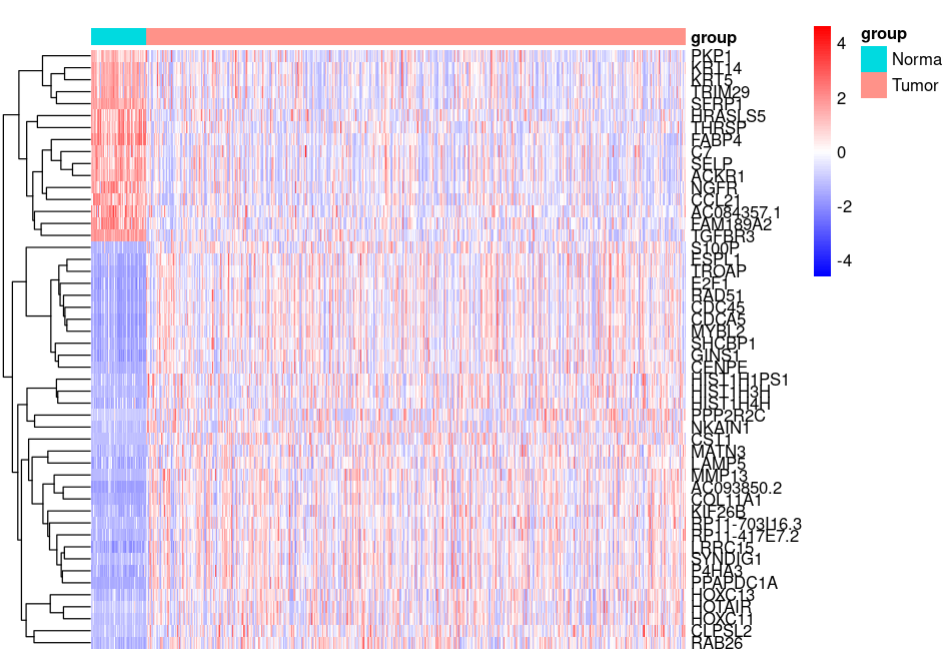
data_deg <- subset(data, regulate %in% c("down-regulated", "up-regulated"))
TCGA_BRCA_pheatmap <- TCGA_BRCA[data_deg$Genes,]
annodata <- data.frame("sample" = colnames(TCGA_BRCA), "group" = group)
rownames(annodata) <- colnames(TCGA_BRCA)
annodata <-annodata[order(annodata$group),]
annodata <- annodata[,-1, drop = FALSE]
color<- colorRampPalette(c('blue','white','red'))(100)
n=t(scale(t(TCGA_BRCA_pheatmap)))
n[n==0] <- NA
n[is.na(n)] <- min(n,na.rm = T)*0.01
mads = apply(n, 1, mad)
m = n[tail(order(mads),50),]
m <- m[, rownames(annodata)]
library(pheatmap)
pheatmap(m , scale = "row",
show_rownames =T,show_colnames = F,
cluster_rows = T,cluster_cols = F,
annotation_col = annodata,
color=color,main = "")
今天的分享就到此,我们后续会对差异基因的富集分析GO,KEGG, GSEA等进行介绍!

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









