







大文件:31万个序列(我看的文章里面说一小时处理100条序列,这里我用了半个小时)
代码如下:把mulfa.fasta文件拆分成单个fasta文件
awk '{if($0~/^>/)a=$0; else{system("echo \\"a"\"\n\""$0a)}}' mulfa.fastamulfa.fasta文件如下:

生成的单个fasta文件如下:

小文件处理及代码解释如这篇文章所说:http://t.csdn.cn/xTZId
感谢这位大佬
使用上述命令生成单个fasta文件后,在linux里很有可能会出出现一个问题,就是文件名最后会多出两个字符“^M”,这是由于windows和linux系统中换行符不一样所导致的,如下图所示,正确的id名后显示一个问号,使用tab键补齐时显示^M。
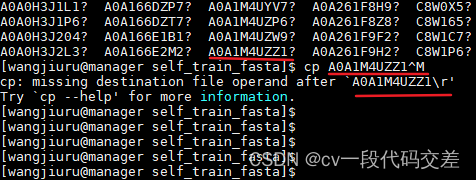
参考这位博主的文章:http://t.csdn.cn/sxPXK
在单个fasta文件夹下,运行下述代码,即可。
#!/bin/sh
fall=$(du -a|awk '{print $2}')
count=0
for dirfile in $fall;
do
count=$(($count+1))
if [ -d $dirfile ]; then
dirfile2=$dirfile/
for ffile in $(ls $dirfile);
do
ffile2=$ffile
mm=$(echo $ffile2|tr -d "\r")
if [ "$ffile" != "$mm" ];then
mv $dirfile2$ffile $dirfile2$mm;
fi
done
fi
done













 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









