







 本文介绍了如何利用ComplexHeatmap包的pheatmap函数绘制热图,并展示了如何进行行列注释、自定义截断和添加额外信息,如平均表达量。通过设置行和列的分组,实现了更个性化的热图展示。此外,还提到了如何结合多个热图以及添加平均值列。ComplexHeatmap提供了丰富的功能,是pheatmap的增强版,适合复杂的热图绘制需求。
本文介绍了如何利用ComplexHeatmap包的pheatmap函数绘制热图,并展示了如何进行行列注释、自定义截断和添加额外信息,如平均表达量。通过设置行和列的分组,实现了更个性化的热图展示。此外,还提到了如何结合多个热图以及添加平均值列。ComplexHeatmap提供了丰富的功能,是pheatmap的增强版,适合复杂的热图绘制需求。
之前我们交替介绍了pheatmap和ComplexHeatmap的热图画法,ComplexHeatmap其实是pheatmap的升级版,包含其大多数功能。小编觉得掌握这两个包就可以解决大多数热图问题了,当然了,很多功能我们也没有涉及,因为太细节化了,需要大家自己在实践中探究学习,至于其他的个性化热图呈现我们下节再讲!
还是用上节的数据,利用ComplexHeatmap包中的pheatmap函数作图。
-
setwd("D:/生物信息学") A <- read.csv("行列注释.csv",header = T,row.names = 1) library(ComplexHeatmap) A <- as.matrix(A) #将表达矩阵转化为matrix pheatmap(A,scale = "row")
简单画一个热图,虽然看似函数很像之前讲的,但是实质不一样,这个是基于ComplexHeatmap包的。
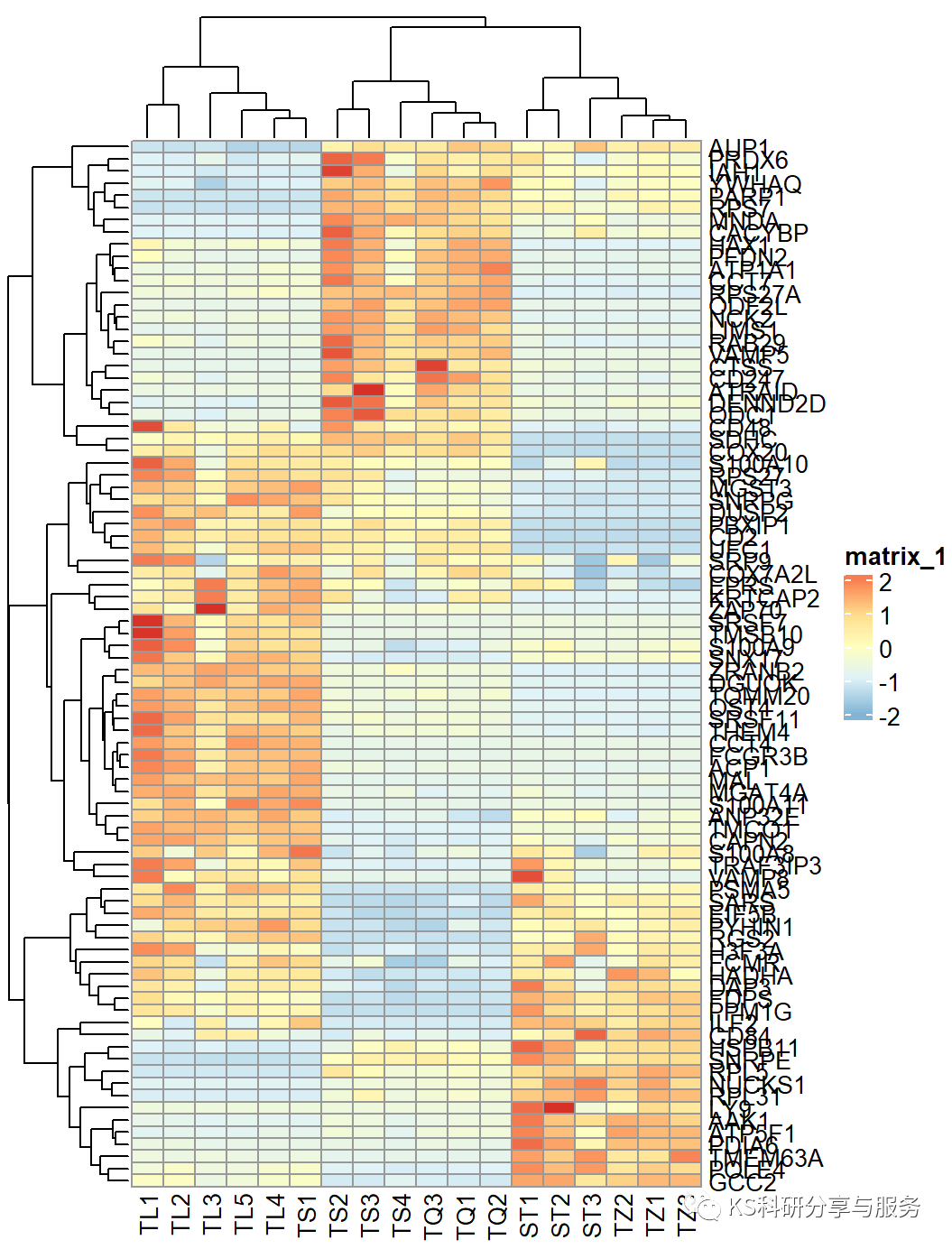
之后的行列信息注释与之前所讲一样,相同的操作手法。
-
annotation_col = data.frame( group = c(rep("ST",3),rep("TZ",3),rep("TL",5),rep("TS",4),rep("TQ",3)), Stage = c(rep("Stage0", 3), rep("Stage1",8), rep("Stage2", 4), rep("Stage3",3)), Age = c(rep("30",2),rep("35",2),rep("30",4),rep("45",3),rep("34",3),rep("33",2),rep("31",2)), Sex = c(rep("F",3),rep("M",3),rep("F",6),rep("M",5),rep("F",1)) ) row.names(annotation_col) <- colnames(A) annotation_row = data.frame( Biological_process = c(rep("Immune response",20), rep("Proteoglycans in cancer", 13), rep("Glycolysis",18),rep("Endocytosis",35)), Pathway = c(rep("Wnt",20), rep("Inflammatory",32),rep("HIF",34)) ) row.names(annotation_row) <- rownames(A)
然后作图,与之前所讲不同之处在于热图截断时不是靠随机的聚类,而是可以自己根据分组截断,更加人性化。
![]()
B <- pheatmap(A, scale = "row",show_rownames = F,#不显示行名
show_colnames = F,#不显示列名
col = colorRampPalette(c("navy","white","firebrick3"))(100),
annotation_col = annotation_col, #列注释信息
annotation_row = annotation_row,#行注释信息
row_split = annotation_row$Pathway,#行截断(按照pathway,不像之前随机)
column_split = annotation_col$group,#列截断
annotation_names_row = F,#不显示行注释信息
annotation_names_col = F ,#不显示列注释信息
column_title = NULL,#不显示列标题
row_title = NULL)#不显示行标题然后还可以复习一下之前讲的,热图只显示感兴趣的基因。
-
genes <- c("S100A8", "ILF2", "RPS27", "HAX1", "PBXIP1", "KRTCAP2", "FDPS", "DAP3", "TRAF3IP3", "EPRS", "CAPN2", "SRP9", "VAMP8", "VAMP5", "MAL", "DUSP2", "ZAP70") genes <- as.data.frame(genes) B + rowAnnotation(link = anno_mark(at = which(rownames(A) %in% genes$genes), labels = genes$genes, labels_gp = gpar(fontsize = 10)))
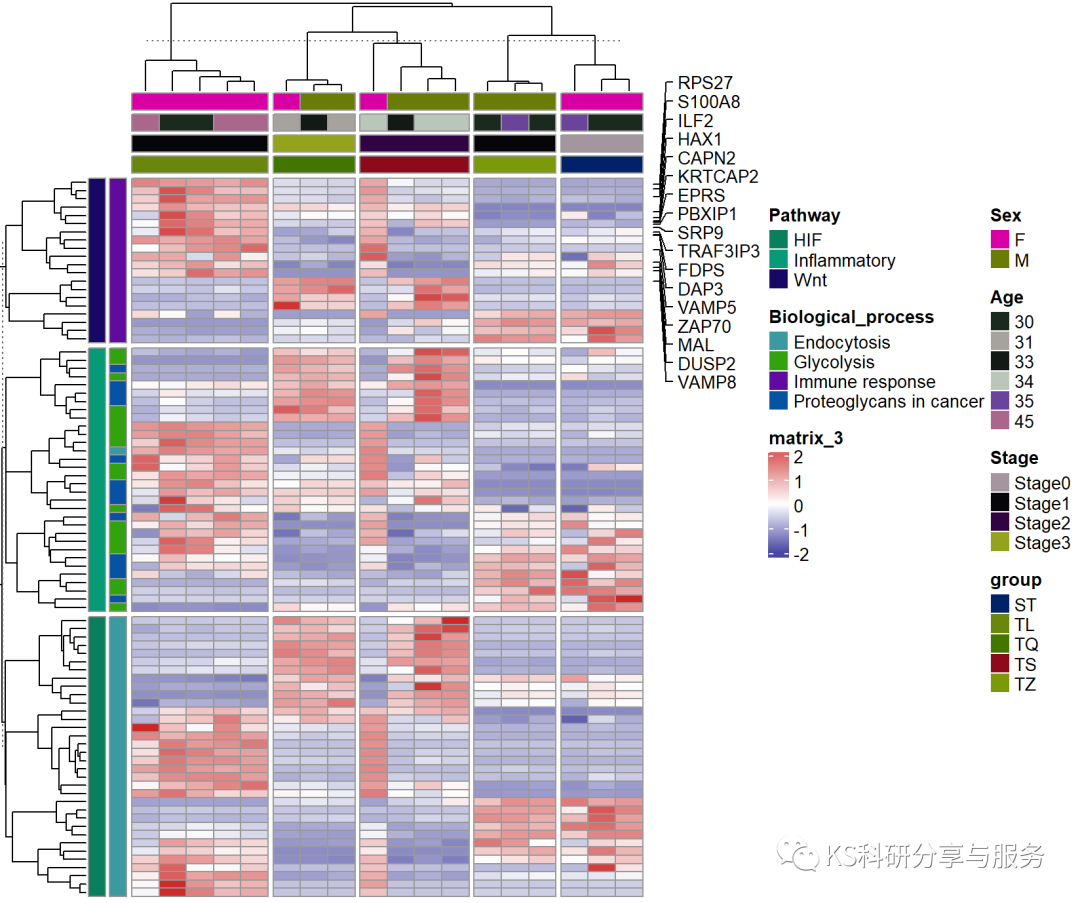
ComplexHeatmap包随机出现的染色搭配小编觉得都还可以,所以不再修饰颜色,修饰的方法和之前一样。其实到这里我们注意到每画一次热图就会随机设置一种颜色,所以为了保持一致,最好设置随机种子(seed())。
如果有两个热图(B和C)想要在一起排列,可以利用如下非常简单的代码:
B + C
到这里还没有结束,这个热图还可以更加复杂,在其行列再加入一些信息,这里举个例子,具体按照具体情况设置。假设我们要展示每个基因的平均表达量,并将其在热图体现,就可以这么做,在原来的热图上加一列:
-
baseMean = rowMeans(A)#求个平均值 pheatmap(A, scale = "row",show_rownames = F, show_colnames = F, col = colorRampPalette(c("navy","white","firebrick3"))(100), annotation_col = annotation_col, annotation_row = annotation_row, row_split = annotation_row$Pathway, column_split = annotation_col$group, annotation_names_row = F, annotation_names_col = F , column_title = NULL, row_title = NULL)+ Heatmap(baseMean,show_row_names = F)
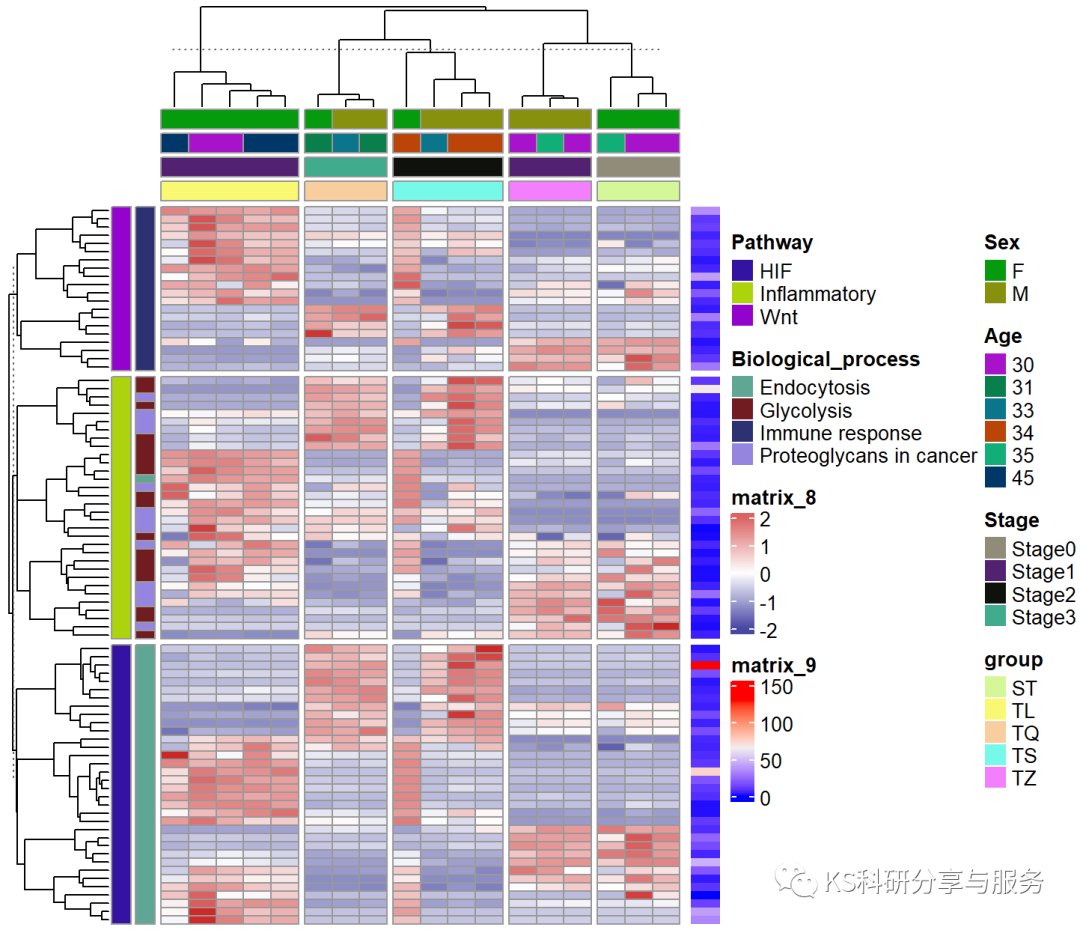
ComplexHeatmap还有超级多的功能,有兴趣可以去探索,看看作者原文。这里讲的已经够用了,没必要把所有的都学会。不过如果后面在文献中见到其他有趣的图,我们还是会去复现,讲解做法的!
记得点赞+关注,学习不迷路!!!
更多精彩内容请访问《KS科研分享与服务》公众号

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









