






这篇帖子的前身可以追溯到:玩转单细胞(2):Seurat批量做图修饰。当时有人问了一个问题,可以添加显著性吗?我们的回答是你需要提取相关组的表达量,进行检验后再用ggplot函数添加即可;或者直接提取数据用ggplot作图那么显著添加也就不成问题了。时隔3月,我们这里提供 了一种函数,可以进行基因在两组之间的显著性分析。同时可进行批量的基因分析。并输出dataframe结果。同时直接在Vlnplot下循环添加显著性。但缺点是只能进行两组比较分析。完整代码已上传群文件! 一般的seurat小提琴图绘制:
library(Seurat)
library(ggplot2)
library(ggpubr)
library(dplyr)
VlnPlot(mouse_data, features = 'S100a8', group.by = 'orig.ident')+
theme_classic() +
theme(axis.text.x = element_text(size = 10,color="black"),
axis.text.y = element_text(size = 10,color="black"),
axis.title.y= element_text(size=12,color="black"),
axis.title.x = element_blank(),
legend.position='none')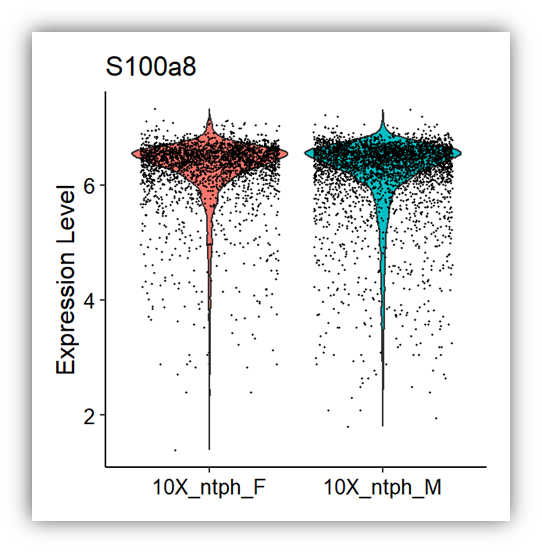
显著性检验函数,有点长,可自行保存成R文件,然后每次使用的时候source一下就可以了。
singlecell_gene_test <- function(SerautObj,
genes.use,
group.by=NULL,
assay = "RNA",
comp = NULL,
alpha_start = .05,
Bonferroni = T,
only_postive =F) {
p_val.out <- c()
stat.out <- c()
condition.out <- c()
gene.out <- c()
if (only_postive == F){
for (gene in genes.use){
group1_cellname = rownames(SerautObj@meta.data[SerautObj@meta.data[[group.by]] == comp[1],])
group1_exp = SerautObj@assays[[assay]]@data[gene, group1_cellname]
group2_cellname = rownames(SerautObj@meta.data[SerautObj@meta.data[[group.by]] == comp[2],])
group2_exp = SerautObj@assays[[assay]]@data[gene, group2_cellname]
t_out = t.test(group1_exp, group2_exp)
cond = paste(comp[1], comp[2], sep = "_")
condition.out <- c(condition.out, cond)
stat.out <- c(stat.out, t_out[["statistic"]])
p_val.out <- c(p_val.out, t_out[["p.value"]])
gene.out <- c(gene.out, gene)
}
}
else{
for (gene in genes.use){
group1_cellname = rownames(SerautObj@meta.data[SerautObj@meta.data[[group.by]] == comp[1],])
group1_exp = SerautObj@assays[[assay]]@data[gene, group1_cellname]
group1_exp <- group1_exp[which(group1_exp>0)]
group2_cellname = rownames(SerautObj@meta.data[SerautObj@meta.data[[group.by]] == comp[2],])
group2_exp = SerautObj@assays[[assay]]@data[gene, group2_cellname]
group2_exp <- group2_exp[which(group2_exp>0)]
t_out = t.test(group1_exp, group2_exp)
cond = paste(comp[1], comp[2], sep = "_")
condition.out <- c(condition.out, cond)
stat.out <- c(stat.out, t_out[["statistic"]])
p_val.out <- c(p_val.out, t_out[["p.value"]])
gene.out <- c(gene.out, gene)
}
}
if (Bonferroni == T){
new_alpha = alpha_start/(2*length(genes.use))
cat(paste("\n", "P-value for significance: p <", new_alpha, "\n"))
sig_out = p_val.out < new_alpha
dfOUT<- data.frame(gene=gene.out, condition = condition.out, p_val = p_val.out, statistic = stat.out, significant = sig_out)
dfOUT$sig = ifelse(dfOUT$p_val > 0.05, "ns",
ifelse(dfOUT$p_val > 0.01, '*',
ifelse(dfOUT$p_val > 0.001, "**", "****")))
}
else{
dfOUT<- data.frame(gene=gene.out, condition = condition.out, p_val = p_val.out, statistic = stat.out)
dfOUT$sig = ifelse(dfOUT$p_val > 0.05, "ns",
ifelse(dfOUT$p_val > 0.01, '*',
ifelse(dfOUT$p_val > 0.001, "**", "****")))
}
return(dfOUT)
}显著性检验:
A <- singlecell_gene_test(mouse_data,
genes.use = c('S100a8','Ltf','Ncf1','Ly6g','Anxa1','Il1b'),
group.by = 'orig.ident',
comp = c("10X_ntph_F", "10X_ntph_M"))
A1 <- singlecell_gene_test(mouse_data,
genes.use = c('S100a8','Ltf','Ncf1','Ly6g','Anxa1','Il1b'),
group.by = 'orig.ident',
comp = c("10X_ntph_F", "10X_ntph_M"),
only_postive = T)作图即可:
anno_pvalue <- format(A$p_val, scientific = T,digits = 3)
anno_sig <- A$sig
plots_violins <- VlnPlot(mouse_data,
cols = c("limegreen", "navy"),
pt.size = 0,
group.by = "orig.ident",
features = c('S100a8','Ltf','Ncf1','Ly6g','Anxa1','Il1b'),
ncol = 3,
log = FALSE,
combine = FALSE)
for(i in 1:length(plots_violins)) {
data <- plots_violins[[i]]$data
colnames(data)[1] <- 'gene'
plots_violins[[i]] <- plots_violins[[i]] +
theme_classic() +
theme(axis.text.x = element_text(size = 10,color="black"),
axis.text.y = element_text(size = 10,color="black"),
axis.title.y= element_text(size=12,color="black"),
axis.title.x = element_blank(),
legend.position='none')+
scale_y_continuous(expand = expansion(mult = c(0.05, 0.1)))+
scale_x_discrete(labels = c("Female","Male"))+
geom_signif(annotations = anno_sig[i],
y_position = max(data$gene)+0.5,
xmin = 1,
xmax = 2,
tip_length = 0)
}
CombinePlots(plots_violins)
或者添加p值:
plots_violins <- VlnPlot(mouse_data,
cols = c("limegreen", "navy"),
pt.size = 0,
group.by = "orig.ident",
features = c('S100a8','Ltf','Ncf1','Ly6g','Anxa1','Il1b'),
ncol = 3,
log = FALSE,
combine = FALSE)
for(i in 1:length(plots_violins)) {
data <- plots_violins[[i]]$data
colnames(data)[1] <- 'gene'
plots_violins[[i]] <- plots_violins[[i]] +
theme_classic() +
theme(axis.text.x = element_text(size = 10,color="black"),
axis.text.y = element_text(size = 10,color="black"),
axis.title.y= element_text(size=12,color="black"),
axis.title.x = element_blank(),
legend.position='none')+
scale_y_continuous(expand = expansion(mult = c(0.05, 0.1)))+
scale_x_discrete(labels = c("Female","Male"))+
geom_signif(annotations = anno_sig[i],
y_position = max(data$gene)+0.5,
xmin = 1,
xmax = 2,
tip_length = 0)
}
CombinePlots(plots_violins)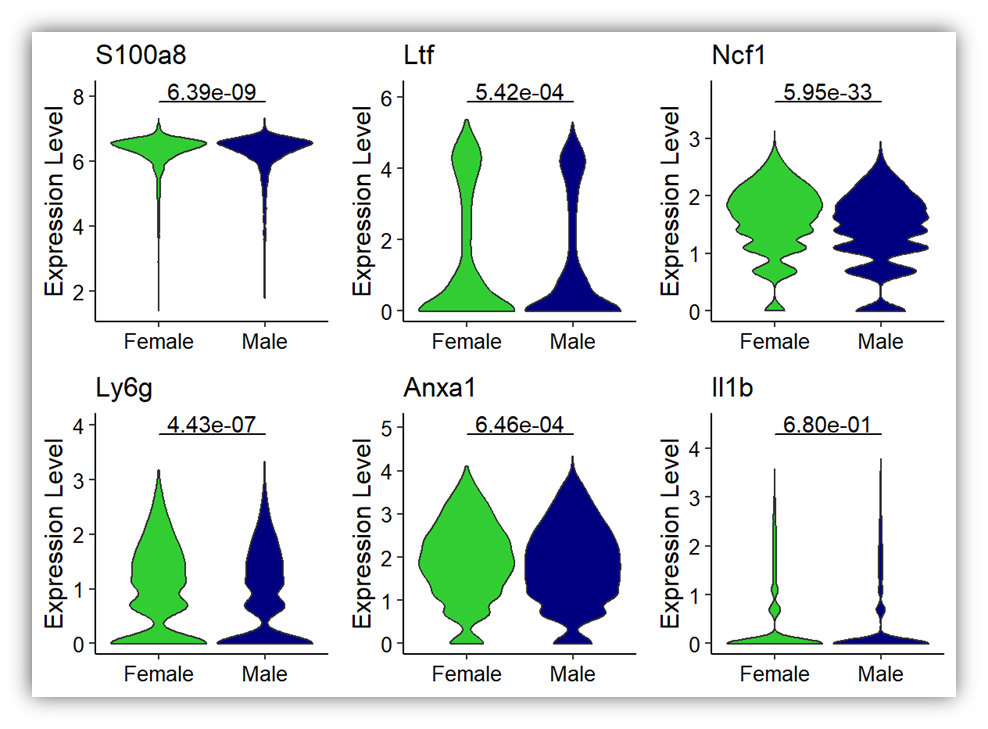
好了。这就是所有内容了,其实这样检验你用不用得到倒是其次,主要是这里面包含一些小的细节知识点,学会了就能和其他内容融汇贯通了,自己感悟吧!更多精彩内容请至KS科研分享与服务公众号















 1100
1100











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









