






20230728
在完成vasp计算所需的四个基本文件(POSCAR/KPOINTS/POTCAR/INCAR/)后,在超算平台上提交作业。
以北京超算云计算中心平台为例,将基本文件放在同一个文件夹里,运行超算平台管理员提供的提交脚本即可提交计算。北京超算云计算中心的提交脚本内容如下:
#!/bin/bash
#SBATCH -p amd_256
#SBATCH -N 2
#SBATCH -n 128
source /public1/soft/modules/module.sh
module load mpi/intel/17.0.7-thc
export PATH=/public1/home/scb6401/software-scb6401/amd-6.3.2:$PATH
srun vasp_std
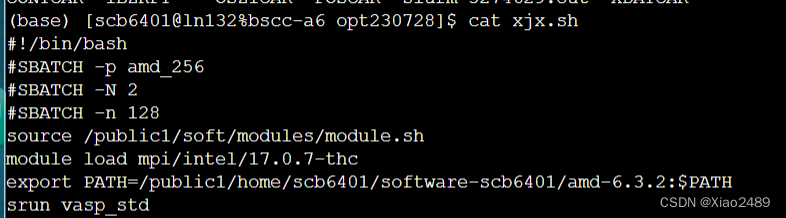
其中,-N 后的数字为使用的节点个数,-n后的为使用的总核数,可以根据实际计算情况在计算前进行修改,北京超算云中心一个节点共有64个核,本脚本使用了两个节点共128个核。使用sbatch xjx.sh即可提交当前目录下的作业。
计算开始后可以用squeue查看当前队列,计算开始后,目录内会出现多个文件,其中 slurm开头的是计算所有的输出结果汇总文件,计算中可以使用tail -f 文件名进行实时结果的查看(crtl+C退出)。使用scancel 任务号可以停止任务的计算。OSZICAR中的数据为计算中出现的所有关于能量的数据。包括计算中所有电子步和离子步的信息。
若计算成功完成,达到INCAR中的收敛条件,且结构不崩溃,则可将CONCAR文件导出用vesta打开即为计算后的结构文件。
优化的INCAR设置:
ISTART=0
ICHARG=2
PREC=M
ISPIN=1
LREAL=A
ALGO=N
NELM=200
EDIFF=1E-4
ENCUT=420
NCORE=16
IVDW=11
IBRION=3
NSW=200
POTIM=0.1
ISIF=2
ISYM=0
EDIFFG=-0.08
ISMEAR=0
SIGMA=0.1
采用分步进行优化,INCAR中ISIF=3,ISYM=0,计算应力张量,取消对称性,进行充分弛豫,不仅弛豫离子位置,同时弛豫晶胞的体积和形状。
将CONCAR中非对角元的非零元素改成0确保c轴垂直于a,b轴,再ISIF=2,仅弛豫离子的位置,保持对称性。
采用一步弛豫:ISIF=3,ISYM=2,(减小POTIM的步长)对称约束下的弛豫。
VESTA中构成缺陷:删除原子,先将体系的对称性降到P1(EDIT中REMOVE SYMEETRY),再在structure中根据原子的序号进行删除,不能直接在界面上直接delete删除(没有实际删除,只是没有显示)。
20230807
结构优化后,计算原子振动频率来验证结构稳定性。
将CONCAR改成POSCAR
在INCAR里修改标签:
修改标签IBRION=5
修改标签NSW=1
(↑此两个用于做振动频率计算)
增加标签NFREE=2
增加标签POTIM=0.015
提交任务进行计算。
计算完成后 gerp "THz" OUTCAR搜索振动频率
20230808

序号 频率 2派×频率 换算成波数 能量
前21个大于0,后三个应该等于0,但由于DFT的误差,f/i小于0.3THz(或10波数以下)即可接受。
频率信息在OUTCAR文件最后。gerp "THz" OUTCAR















 499
499











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









