






20240601
vmd:
not resname dmf and not water
pbc box -color black -width 1.5
位置限制:
限制文件itp:
用要位置限制的分子的pdb或gro文件用gmx genrestr命令生成对应分子的位置限制itp文件:
gmx genrestr -f 1.pdb -o 1——restrat.itp -fc 1000 1000 1000
(相当于用一个很大的限制势限制分子的运动,导致分子的结构和坐标基本不发生不变化)
选取要限制的组
生成限制分子的itp文件
对于模拟流程,先正常做一个能量极小化,再用做一个位置限制NPT模拟,mdp开头改成define = -DPOSRES,然后把
#ifdef POSRES
#include "c6_restra.itp"
#endif
加在需要限制的分子的itp文件的末尾
然后再同样进行位置限制的正常模拟。
模拟中提示势能太大,模拟无法进行:
可能是盒子定的太小,用packmol创建一个更大的盒子,先做一次em看看势能的大小。
模拟中提示盒子的变化超过1%:
可能是初始结果过于不合理,参考上一步,或将分子模拟的步长改成1fs或tau_p改大也可以缓解这种情况。
在NPT过程中出现水分子团聚,盒子内出现很多真空,分子分布不均匀的现象,是因为模拟时长不够,增长模拟时长,即可解决。
或者:把压浴时间常数设小、让压浴作用效果表现得更显著,也可以暂时性加大可压缩系数,以尽可能在较短时间内令盒子尺寸收缩合适。等真空区消差不多了,再改成原本的压浴参数。
在使用位置限制时,预处理时命令需要加-r .gro/.pdb文件,直接加前面的gro文件就行。
wfn文件产生:
在gaussian几何优化时,在命令末尾加上out=wfn,然后在最后加上输出的路径public3/1.wfn,在计算的最后就会将wfn文件自动保存到路径。
或者将chk文件转化为fchk文件后,用Multiwfn的100的2功能,转化格式。
IGMH分析:
可以使用模拟后的.gro文件,用Multiwfn打开,选择20-11,然后选择片段,比如将分子和离子分成不同的片段,输入片段数和不同片段的原子序号,再选择网格形成的方法,
二面角分析:
将结构文件导入VMD,按数字键1再点击原子即可显示序号。
找到两个平面对应的原子序号,将结果文件.gro.xtc导入VMD并保存为.xyz格式。
启动Multiwfn,然后输入
C18dimer.xyz
1000 //隐藏的主功能
201 //隐藏的子功能
1 //获得片段间几何中心距离和夹角
4001 //轨迹总帧数
1-18 //第一个片段里的原子序号,即第一个18碳环
19-36 //第二个片段里的原子序号,即第二个18碳环
瞬间就算完了。在当前目录下出现了distangle.txt,每一列的内容是
第1列:帧号
第2列:两个片段的几何中心间的距离(埃)
第3列:第一个片段平面的法矢量与两个片段几何中心连线之间的夹角,姑且可以叫错位角,体现相对错位程度,示意图见下
第4列:两个片段的法矢量之间的夹角,即一般意义上的平面间夹角。
aNCI分析:
使用Multiwfn做aNCI分析图形化考察动态过程中的蛋白-配体间的相互作用 - 思想家公社的门口:量子化学·分子模拟·二次元
将生成的轨迹文件和结构文件去掉水。
选择语句在VMD里用的地方非常多,无处不在。比如如果想在图形窗口只显示指定的区域,那么可以在Graphics - Representation里在selected atoms的地方写上选择语句。如果想把指定的区域保存成新文件,那么可以在File - Save coordinates里在Selected atoms写上选择语句。
xtc载入前删除gro文件的一帧再载入。
然后not water保存轨迹文件和结构文件。
把轨迹切换到第0帧,然后在Graphics - Representation里把选择范围设置为resname MOL or same resid as within 3.5 of resname MOL,这样就把配体以及有任何一个原子在距离配体3.5埃范围以内的分子都选中。把显示方式设为CPK。
CPK模式:
导入前输入设置默认状态
mol default style {CPK 1.0 0.3 30 30}再导入即可。
把cluster.xyz和cluster.pdb放到相同目录下,然后启动Multiwfn,输入cluster.xyz的路径,程序载入cluster.xyz里的坐标时也会把cluster.pdb里的元素信息载入。
20 //图形化分析弱相互作用
3 //aNCI分析
1,1001 //考虑的轨迹帧号范围,从1到1001帧(注意Multiwfn的帧号从0开始计)
11 //设置格点。这里选的模式11专门适合aNCI分析,即选择一批原子,然后往周围扩展一定距离定义盒子范围
144-161 //配体的原子序号(打开cluster.pdb,可见MOL残基第一个原子和最后一个原子的原子序号分别是144和161)
3 A //往配体周围扩展3埃定义盒子
0.15 //格点间距。数值越小要算的点数越多,耗时越高,而aNCI等值面会越光滑
之后选6,aRDG(平均的约化密度梯度)格点数据会被导出到当前目录下的avgRDG.cub,平均的sign(lambda2)rho会被导出到当前目录下的avgsl2r.cub。如果还想绘制热波动指数(Thermal fluctuation index, TFI)着色的aRDG等值面图,这里再选7来再计算TFI,之后TFI格点数据会被输出到当前目录下的thermflu.cub。
把avgRDG.cub和avgsl2r.cub以及Multiwfn目录下的examples\aNCI\avgRDG.vmd脚本挪到VMD目录下。然后启动VMD,在文本窗口里输入source avgRDG.vmd执行此脚本绘制aNCI图。
如果出现部分零碎的多余的等值面,需要对aRDG的格点数据进行屏蔽。在Multiwfn中,可以实现将距离某个片段较远区域的aRDG格点数据的数值设为一个比较大的值(比如100),之后再绘制等aRDG值面图的时候那部分区域就不会出现等值面了,下面就来这么处理一下。
这里先把原先avgRDG.cub改名为avgRDG_org.cub,然后启动Multiwfn,载入avgRDG_org.cub,输入
13 //处理格点数据
13 //设置远离或接近某些原子的格点的数值
1.6 //设置原子范德华半径乘的倍数,这个值可以反复试试直到效果满意
100 //满足条件的格点的数值设为100
2 //手动输入原子序号
144-161 //配体的原子序号
0 //将当前的格点数据导出
avgRDG.cub //新的格点数据的文件名
之后将avgRDG.cub挪到VMD目录下替换掉原先的,再次用avgRDG.vmd脚本绘图。之后再做一些适当调节,在Graphics - Representation里把显示等值面的那个rep的isovalue稍微设大到0.3使得等值面更膨胀丰满一些,以令边缘的锯齿减弱。然后把显示当前几何结构的rep的选择范围设为serial 144 to 161,把Sphere Scale设小为0.7。然后点Create Rep按钮新建一个Rep,把选择范围设为not serial 144 to 161,Drawing Method设为licorice,bond radius设0.1,Material设Transparent。此时的图像效果如下。
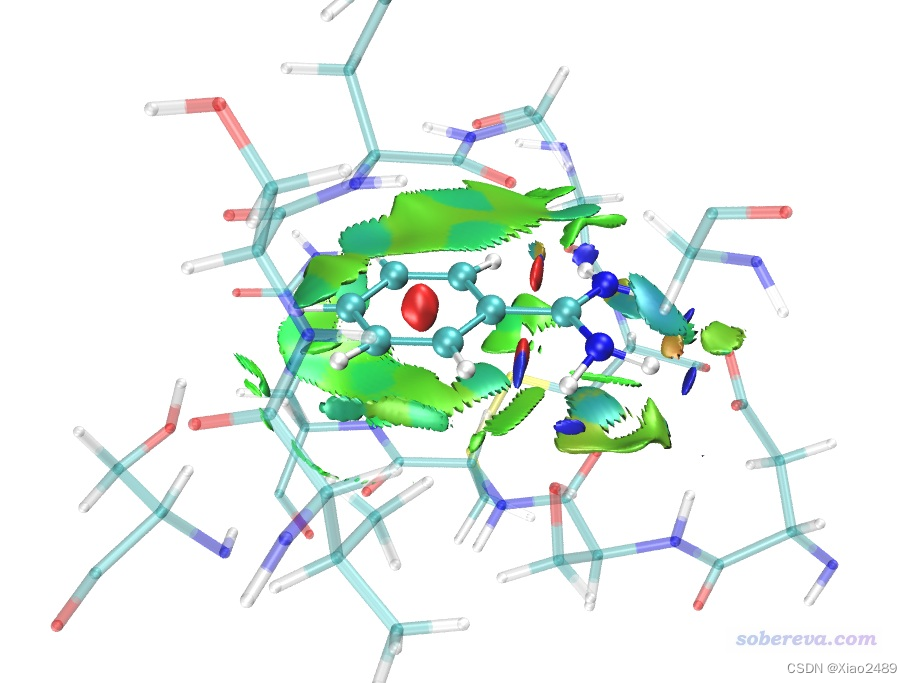
绘制静电势:
使用Multiwfn+VMD快速地绘制静电势着色的分子范德华表面图和分子间穿透图(含视频演示)
用chg文件分析静电势,然后复制到vmd文件夹,用iso绘图,定量分析chg的静电势(带电体系必须要改),然后根据静电势范围设置vmd的范围即可。
计算原子间距离
先用win生成ndx文件
gmx make_ndx -f md.gro -o index.ndx
之后选择需要的原子或者分子组成组,最后q保存退出。
gmx distance -s md.tpr -f fixedmd-md.xtc -n index.ndx -oav 1.xvg
然后输入需要输出的两个组的组名即可
安装qtgrace画出1.xvg















 2696
2696

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









