







1. Introduction
随着大量生物医学数据和知识的收集与利用以及在许多应用领域取得巨大成功的深度学习技术的进步,药物发现过程,特别是DTI预测得到了显著增强。最近,各种深度模型在DTI预测中显示出令人鼓舞的表现。它们通常将药物和蛋白质数据作为输入,将DTI视为一个分类问题,并通过深度学习模型对输入进行预测。尽管做出了这些努力,一下挑战仍然存在:
- 交互机制建模不足。现有的研究学习分子表征并基于药物和蛋白质的整个分子结构进行预测,忽略了相互作用是只涉及药物和蛋白质的相关子结构的子结构。全结构分子表征引入噪声并影响预测性能。此外,学习到的表征很难解释,因为它们不能提供一条易于理解的路径来指示药物和蛋白质的哪些子结构有助于相互作用。
- 限于有限的标记数据。先前的工作仅关注手头的数据,将范围限制在数千种药物和蛋白质中,而忽略了大量未标记的生物医学数据。
为了解决这些挑战,我们提出了一种基于 transformer 的生物启发分子数据表征方法(MolTrans)来利用大量未标记数据进行 DTI 预测。做出了以下贡献:
-
知识启发的表征和交互建模,以实现更准确、更可解释的预测。受 DTI 是子结构的知识启发,MolTrans 提出了频繁连续子序列(FCS)挖掘的数据驱动方法,该方法适用于提取蛋白质和药物的高质量且尺寸合适的子结构。此外,MolTrans 还包括模拟真实生物DTI过程的生物启发交互模块。新的子结构识别使得能够通过交互模块中的显式映射来理解哪个子结构组合与结果更相关。
-
利用大量未标记的生物医学数据。MolTrans 挖掘来自多个未标记数据源的数百万药物和蛋白质序列以提取药物和蛋白质的高质量子结构。与单独使用小的训练数据集相比,海量数据产生了更高质量的子结构。我们还使用 Transformer 来增强特征,该 Transformer 从未标记数据生成的大序列子结构输出中捕获复杂信号。
我们提供了各种真实药物发现设置(包括未发现的药物/靶点问题)和稀缺训练数据集设置中最先进方法的综合性能比较。实验结果证明,MolTrans 的预测性能比最先进的基线水平提高了25%。MolTrans 与现有模型的不同之处在于:(i)它是基于知识驱动的模型架构而不是直接应用现有的深度学习模型;(ii)强调可解释性而不是预测性能;(iii)使用外部药物和靶点数据来补充相互作用数据集。
2. Materials and methods
2.1 Problem definition
我们将 DTI 预测作为一个分类任务来确定一对药物和靶蛋白是否会相互作用。在我们的设置中,药物
i
i
i 由SMILES字符串
S
i
S_i
Si 表示。我们用
S
S
S 表示药物的SMILES表征,用
A
A
A 表示目标蛋白(由一系列氨基酸token表示)。DTI 预测任务定义如下:
给定
n
n
n 种药物序列
S
=
{
S
1
,
S
2
,
⋯
,
S
n
}
S=\{S_1,S_2,\cdots,S_n\}
S={S1,S2,⋯,Sn} 和
m
m
m 个蛋白质序列
A
=
{
A
1
,
A
2
,
⋯
,
A
m
}
A=\{A_1,A_2,\cdots,A_m\}
A={A1,A2,⋯,Am},学习一个从药物-靶点对到相互作用概率分数的映射函数
F
:
S
×
A
→
[
0
,
1
]
F:S\times A\rightarrow[0,1]
F:S×A→[0,1]
2.2 The MolTrans method
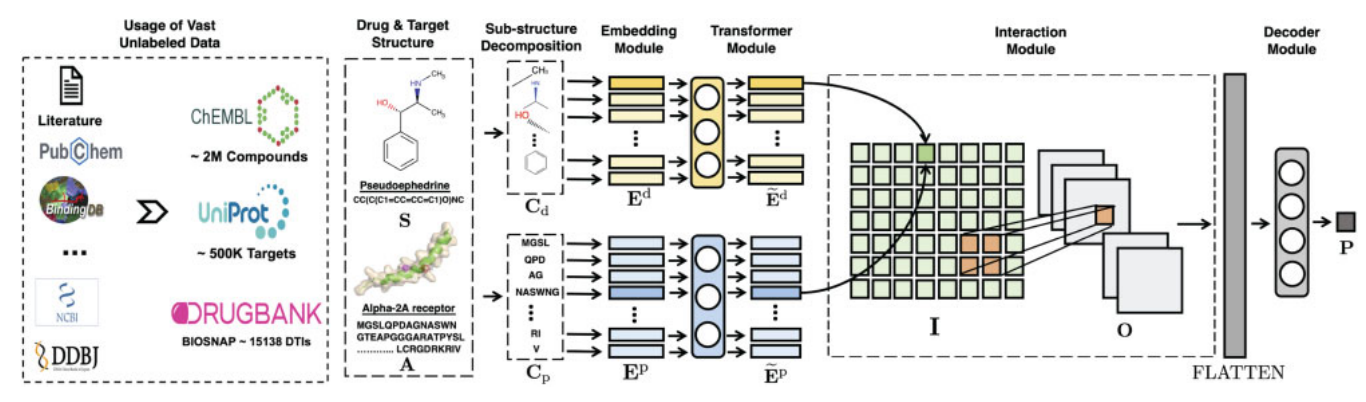
给定输入的药物和蛋白质数据,FCS挖掘模块首先使用专门的分解算法将其分解为一组明确的子结构序列,然后将输出送入一个增强的 Transformer 嵌入模块中来获得每个子结构的增强的上下文嵌入,接下来在相互作用预测模块中,用成对相互作用分数来将药物子结构和蛋白质子结构配对,随后在相互作用图上用 CNN 来捕获高阶相互作用,最后用解码器模块输出概率分数。
2.2.1 FCS mining module
在子结构级别 DTI 的领域知识驱动下,MolTrans 首先将蛋白质和药物的分子序列分解为子结构。特别是,我们提出了一种称为 FCS 算法的数据驱动的序列模式挖掘算法用来发现药物和蛋白质数据库中的重复子序列。受自然语言处理领域子词单元的启发,FCS 旨在为序列生成一组频繁子序列的层次结构。

FCS 将蛋白质/药物的每个序列分层分解为子序列、更小的子序列和单个原子/氨基酸符号。
FCS 首先初始化一个由特定的氨基酸 token 或 SMILES 字符串字符和给定 token 组成的集合 V V V,标记整个药物/蛋白质语料库得到集合 W W W。然后,扫描 W W W 并找到最频繁的连续标记 ( A , B ) (A,B) (A,B),FCS 用 ( A B ) (AB) (AB) 来更新集合 W W W 中的 ( A , B ) (A,B) (A,B),并将新 token 添加到集合 V V V 中。然后重复扫描、识别和更新过程,直到没有频繁标记超过阈值 θ \theta θ 或 V V V 的大小达到了预定义的最大值 l l l。
通过此操作,足够频繁的子序列被合并为一个 token,而不够频繁的子序列被分解为一组较短的 token。最终,对于每个药物/蛋白质,FCS产生大小为
k
k
k 的药物/靶点子结构序列
C
=
{
C
1
,
C
2
,
⋯
,
C
k
}
C=\{C_1,C_2,\cdots,C_k\}
C={C1,C2,⋯,Ck},其中
C
i
C_i
Ci 来自集合
V
V
V。
使用 FCS 算法,MolTrans 将输入药物和靶点分别转换成一系列显示子结构
C
d
C_d
Cd 和
C
p
C_p
Cp。它有如下两点意义:
- 与以前的子结构识别方法相比其更容易解释。FCS药物编码能够提供明确的提示,因为它将每个药物分子分解为离散的中等大小的子结构分区。它允许利用大量未标记的数据来改进子结构挖掘。例如,我们使用的 Uniprot 数据集由560 823个独特的蛋白质序列组成,ChEMBL数据库包括1870 461个药物SMILES字符串。我们观察到,挖掘的子结构的质量来源于我们使用的大量未标记数据。在小数据集中,许多有用的子结构的出现频率低于合理的最小频率,而大的聚集数据集可以用更大的序列池成功地识别它们。
- 可以捕获基本且有意义的生物医学语义。产生的子结构与药物和蛋白质频繁出现的基本单位有关。我们发现,在给定不同数据集特征(如蛋白质数据集的不同生物类型和药物数据集的药物相似性)的情况下,FCS算法识别了相似的基本生化子结构集,这表明FCS算法的鲁棒性。通常,我们应用更通用的数据集。
2.2.1 Augmented transformer embedding module
为了捕获子结构的化学语义,MolTrans 包括一个增强嵌入模块(它首先初始化一个可学习的子结构查找字典,然后通过 transformer 编码器用上下文子结构信息增强嵌入)。我们用 transformer 进行分子表征学习。在我们的设置中,transformer 编码器中的自注意力机制通过学习来自同一分子的所有子结构来修改每个输入子结构嵌入。这样得到的子结构嵌入更好,因为它通过考虑相邻子结构之间的复杂化学关系而具有上下文意义。
具体而言,对于每个输入药物-靶点对,我们将相应的子结构序列
C
p
C_p
Cp 和
C
d
C_d
Cd 转换为两个矩阵
M
p
∈
R
k
×
θ
p
M^p\in R^{k\times \theta_p}
Mp∈Rk×θp 和
M
d
∈
R
l
×
θ
d
M^d\in R^{l\times \theta_d}
Md∈Rl×θd,其中
k
/
l
k/l
k/l 是药物/蛋白质子结构的总大小,
θ
p
\theta_p
θp 和
θ
d
\theta_d
θd 是蛋白质和药物的子结构序列的最大长度,并且每列
M
i
p
M^p_i
Mip 和
M
j
d
M^d_j
Mjd 是对应于蛋白质序列的第
i
i
i 个子结构和药物序列的第
j
j
j 个子结构的子结构索引的一个 one-hot 向量。每个蛋白质和药物的上下文嵌入
E
c
o
n
t
p
E_{cont}^p
Econtp 和
E
c
o
n
t
d
E_{cont}^d
Econtd 由一个可学习的词汇查找矩阵
W
c
o
n
t
p
∈
R
v
×
k
W^p_{cont}\in R^{v\times k}
Wcontp∈Rv×k 和
W
c
o
n
t
d
∈
R
v
×
l
W^d_{cont}\in R^{v\times l}
Wcontd∈Rv×l 生成:
E
c
o
n
t
i
p
=
W
c
o
n
t
p
M
i
p
,
E
c
o
n
t
j
d
=
W
c
o
n
t
d
M
j
d
E_{cont_i}^p=W^p_{cont}M^p_i,E_{cont_j}^d=W^d_{cont}M^d_j
Econtip=WcontpMip,Econtjd=WcontdMjd其中
v
v
v 每个子结构的潜在嵌入的大小。
由于 MolTrans 使用了序列子结构,我们也包括一个由可学习的词汇查找矩阵
W
p
o
s
p
∈
R
v
×
θ
p
W^p_{pos}\in R^{v\times \theta_p}
Wposp∈Rv×θp 和
W
p
o
s
d
∈
R
v
×
θ
d
W^d_{pos}\in R^{v\times \theta_d}
Wposd∈Rv×θd 生成位置嵌入
E
p
o
s
i
p
E_{pos_i}^p
Eposip 和
E
p
o
s
j
d
E_{pos_j}^d
Eposjd:
E
p
o
s
i
p
=
W
p
o
s
p
I
i
p
,
E
p
o
s
j
d
=
W
p
o
s
d
I
j
d
E_{pos_i}^p=W^p_{pos}I^p_i,E_{pos_j}^d=W^d_{pos}I^d_j
Eposip=WpospIip,Eposjd=WposdIjd其中
I
i
p
∈
R
θ
p
,
I
j
d
∈
R
θ
d
I^p_i\in R^{\theta_p},I^d_j\in R^{\theta_d}
Iip∈Rθp,Ijd∈Rθd 是第
i
i
i 个位置和第
j
j
j 个位置为 1 的 one-hot 向量。
最终的嵌入向量
E
i
p
,
E
j
d
E^p_i,E^d_j
Eip,Ejd 是上下文嵌入和位置嵌入的和:
E
i
p
=
E
c
o
n
t
i
p
+
E
p
o
s
i
p
,
E
j
d
=
E
c
o
n
t
j
d
+
E
p
o
s
j
d
(1)
E^p_i=E^p_{cont_i}+E^p_{pos_i},E^d_j=E^d_{cont_j}+E^d_{pos_j}\tag{1}
Eip=Econtip+Eposip,Ejd=Econtjd+Eposjd(1)上述模型输出一组独立的子结构嵌入。然而,这些子结构之间有化学关系来捕获这些上下文信息,我们进一步使用 transformer 编码器层来增强嵌入:
E
~
p
=
Transformer
Protein
(
E
p
)
,
E
~
d
=
Transformer
Drug
(
E
d
)
(2)
\widetilde{E}^p=\text{Transformer}_{\text{Protein}}(E^p),\widetilde{E}^d=\text{Transformer}_{\text{Drug}}(E^d)\tag{2}
E
p=TransformerProtein(Ep),E
d=TransformerDrug(Ed)(2)
2.2.3 Interaction prediction module
MolTrans 包括一个由两层组成的交互模块:① 一个用于对成对子结构相互作用进行建模的相互作用张量。② 一个用于提取邻域相互作用的相互作用映射的 CNN 层。
成对作用
为了对成对作用进行建模,对于蛋白质中的每个子序列
i
i
i 和药物中的每个子序列
j
j
j,有:
I
i
,
j
=
F
(
E
~
i
p
,
E
~
j
d
)
(3)
I_{i,j}=F(\widetilde{E}_i^p,\widetilde{E}_j^d)\tag{3}
Ii,j=F(E
ip,E
jd)(3)其中
F
F
F 是衡量对间相互作用的函数,可以是和、平均值、点积等。因此,在这个层之后,我们得到一个张量
I
∈
R
θ
d
×
θ
p
×
Φ
I\in R^{\theta_d\times \theta_p\times \Phi}
I∈Rθd×θp×Φ,其中
θ
d
/
θ
p
\theta_d/\theta_p
θd/θp 分别是药物/蛋白质子序列的长度,
Φ
\Phi
Φ 是函数
F
F
F 的输出的大小,这个张量的每列考虑了药物和蛋白质的单个子结构的相互作用。为了提供可解释性,我们倾向于将点积作为聚集函数,因为它生成一个可以明确衡量单个药物-靶点子结构对之间的相互作用强度的标量。由于每对点积输出是一维的,所以
I
I
I 变成了二维作用图。如果图中的某个值很高,它将在下游层中激活并且具有更高的DTI相互作用可能性。通过端到端的学习,如果一对子结构确实相互作用,它们将在相互作用图中相应的子结构对位置上具有较高的相互作用分数。因此,通过检查此图,我们可以直接看到哪些子结构对有助于最终结果。
邻域作用
蛋白质和药物的邻近子结构在触发相互作用时相互影响。因此,除了对单独的成对作用进行建模外,还需要对附近区域的相互作用进行建模。通过作用图
I
I
I 顶部的CNN层来实现这一点。直觉是,通过应用几个有序不变的局部卷积核,附近区域的交互可以被捕获并聚合。最终得到输入药物-靶点对的输出表征
O
O
O:
O
=
C
N
N
(
I
)
(4)
O=CNN(I)\tag{4}
O=CNN(I)(4)该互作用模块的灵感来自深度交互推理网络,由于这种显示互作用建模,我们可以从互作用图中可视化各个子结构互作用对的强度。为了输出指示互作用可能性的概率,我们首先将
O
O
O 展开为向量,并使用由权重矩阵
W
o
W_o
Wo 和偏置向量
b
o
b_o
bo 参数化的线性层:
P
=
σ
(
W
o
⋅
FLATTEN
(
O
)
+
b
o
)
(5)
P=\sigma(W_o·\text{FLATTEN}(O)+b_o)\tag{5}
P=σ(Wo⋅FLATTEN(O)+bo)(5)其中
σ
\sigma
σ 是
sigmoid
\text{sigmoid}
sigmoid 函数。
二分类的损失函数为:
L
=
Y
⋅
log
(
P
)
+
(
1
−
Y
)
⋅
log
(
1
−
P
)
(6)
L=Y·\log(P)+(1-Y)·\log(1-P)\tag{6}
L=Y⋅log(P)+(1−Y)⋅log(1−P)(6)其中
Y
Y
Y 是标签值。
2.3 Implementation
对于 FCS 算法,我们将数据集中药物和蛋白质子结构的最小出现次数设置为 500,得到 23532 个药物子结构和 16693 个蛋白质子结构。对于 transformer 编码器,我们为药物和蛋白质使用两层 transformer 编码器。输入嵌入的大小为 384,我们为每个中间维度为 1536 的 transformer 编码器设置了 12 个注意头。我们将药物的最大序列长度设置为 50,将蛋白质的最大序列长设置为 545,以覆盖数据集中 95% 的序列。对于CNN,我们使用三个内核大小为三的过滤器。对于优化超参数,我们使用学习率为 0.00001 的Adam优化器,batch_size 设置为64,并允许它运行30个 epochs。dropout 设为0.1。
3. Result
我们的实验回答了如下问题:
- Q1:MolTrans 是否提高了 DTI 预测性能?
- Q2:MolTrans 如何处理未知的药物/靶点案例?
- Q3:MolTrans 如何应对大量丢失的数据?
- Q4:不同蛋白质系列的性能如何变化?
- Q5:MolTrans 是否提供了关于 DTI 的有用知识?
- Q6:MolTrans 的各个组件如何对预测性能增益做出贡献?
3.1 Experimental setup
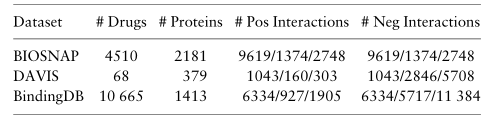
(1)数据集
我们使用 BIOSNAP 收集的 MINER DTI 数据集作为我们的主要实验数据集。它包括 4510 个药物节点和 2181 个蛋白质靶点,以及来自 DrugBank 的 13741 个DTI对。BIOSNAP 数据集仅包含正 DTI 对。对于负 DTI 对,我们按照常见做法从未发现的对中进行采样,获得了具有相等正负样本的平衡数据集。除了BIOSNAP,我们的预测性能比较实验中还包括两个基准数据集。DAVIS 由 68 种药物和 379 种蛋白质的相互作用数据组成,BindingDB 由 10665 种药物和 1413 种蛋白质的互作用数据构成。
(2)指标
我们使用ROC-AUC和PR-AUC作为度量二元分类性能的指标。此外,我们使用敏感性和特异性指标,其中阈值是验证集中F1得分最好的一个。
(3)评估策略
我们以 7:1:2 的比例将数据集分为训练集、验证集和测试集。对于每个实验,我们使用不同的数据集随机拆分进行五次独立运行。然后,我们根据 ROC-AUC 性能从验证集中选择性能最佳的模型。然后,通过验证选择的模型在测试集上进行评估。
3.2 Baselines
我们将 MolTrans 与以下基线进行了比较。我们专注于最先进的深度学习模型,因为它们表现出了优于浅层模型的性能。
- LR:对连接的药物和蛋白质特征向量应用逻辑回归模型。我们对药物的 ECFP4 和 PubChem 以及蛋白质的 PSC 和 CTD 的所有组合进行了实验。我们发现药物的 ECFP4 和蛋白质的 PSC 具有最高的性能。
- DNN:在 ECFP4 和 PSC 连接向量的顶部使用隐藏单元大小为 1024 的三层DNN。
- GNN-CPI:使用图神经网络对药物进行编码,并使用 CNN 对蛋白质进行编码。然后将潜在向量连接到神经网络中用于化合物-蛋白质相互作用预测。我们遵循论文中描述的相同超参数设置。
- DeepDTI:使用 DBN 对 DTI 进行建模,DBN 是一组受限玻尔兹曼机器。它使用 ECFP2、ECFP4、ECFP6 的串联作为药物特征,并使用 PSC 作为蛋白质特征。我们基于验证集性能优化了论文中描述的超参数。
- DeepDTA:将 CNN 应用于原始SMILES字符串和蛋白质序列以提取局部残基模式,任务是预测结合亲和力值。我们在最后添加了一个 Sigmoid 激活函数,将其改为二进制分类问题,并进行超参数搜索以确保公平性。
- DeepConv-DTI:使用CNN和全局最大池层提取蛋白质序列中的各种长度的局部模式,并在ECFP4上应用全连接层。它在不同的数据集上进行了广泛的实验,是 DTI 二进制预测任务中最先进的模型。我们遵循论文中描述的相同超参数设置。
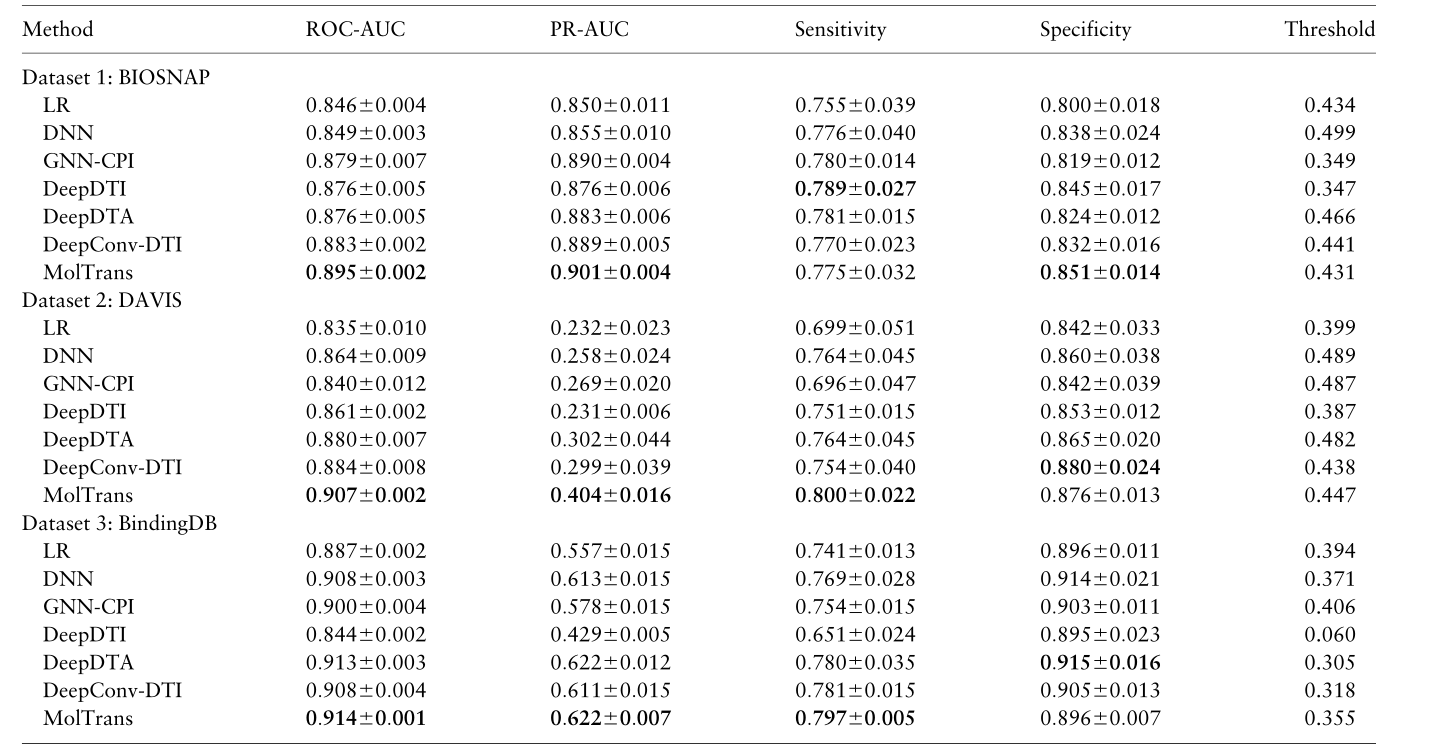
3.3 Q1: MolTrans achieves superior predictive performance
为了回答问题1,我们随机选择20%的药物-靶点对对作为测试集。结果显示,MolTrans 在所有数据集的 ROC-AUC 和 PR-AUC 的 DTI 预测设置中始终具有更好的预测基线。MolTrans 比最佳表现基线(DAVIS PR-AUC)增加了25%。注意,由于不同方法的阈值不同,敏感性和特异性可能不同。
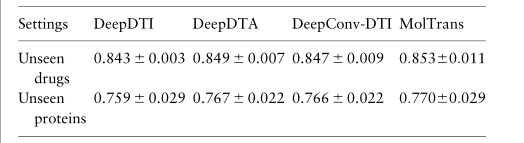
3.4 Q2: MolTrans has competitive performance in unseen drug and target setting
为了模拟未知的药物/靶点任务,我们随机选择 20% 的药物/靶点蛋白以及与这些药物和靶点相关的所有DTI对作为测试集。我们观察到 KronRLS 的性能在不同的设置中有所不同。这是因为 KronRLS 是一种基于相似性的方法;因此,它容易受到手头的数据属性的影响。在未知的药物设置中,我们发现单层 LR 比多层 DNN 更好,并且比具有更复杂深度模型设计的 SOTA 方法更差。这表明了精心设计模型架构的必要性。我们还看到,MolTrans 在这两种情况下都具有与SOTA深度学习基线相比的竞争力。
3.5 Q3: MolTrans performs best with scarce data
我们发现 MolTrans 是最稳健的方法。相反,SOTA基线如 DeepDTI 和 DeepConv DTI 随着缺失分数的增加而下降。MolTrans 在稀缺环境中表现良好的一个原因是,与使用整个药物和蛋白质的其他方法相比,MolTrans 使用了相对丰富的子结构,因此迁移性比较好。
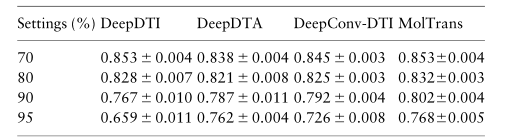
3.6 Q4: MolTrans is robust in various protein families
靶蛋白来自不同的蛋白质家族。重要的是,预测算法不偏向于一个特定的蛋白质家族。在本实验中,我们测试了对四个最大的药物靶点的预测性能:酶、离子通道、G蛋白偶联受体(GPCR)和核受体。我们检索一个 BIOSNAP 测试集,并使用 GtoPdb 数据库将目标蛋白映射到四个蛋白家族。我们发现 1908 个酶相互作用、533 个GPCR相互作用、496 个离子通道相互作用和 104 个核受体相互作用。我们发现 MolTrans 在上述所有单个蛋白质家族中都是稳健的。特别是,酶、GPCR和离子通道具有比总体蛋白质类更高的性能。
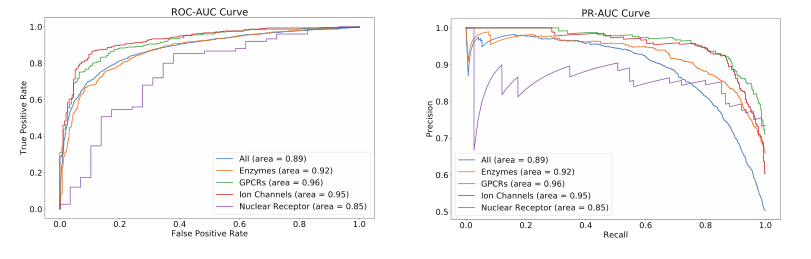
3.7 Q5: MolTrans allows model understanding
相互作用图中的高值细胞代表药物和靶点子结构之间的潜在激活的相互作用,这对最终的相互作用结果很重要。因此,为了可视化,我们生成一张热图,让我看看哪些细胞具有高值。然后,我们选择一个阈值来遮蔽大多数具有低值的细胞。然后我们检查文献,看看剩余的细胞是否包含相互作用结果的线索。
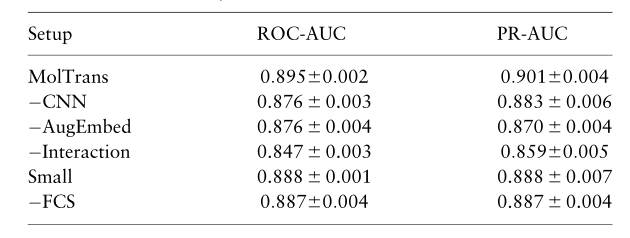
3.8 Q6: Ablation study
我们使用以下设置对全数据设置进行消融研究:
- -CNN:删除了CNN,并展开互作用图 I I I 输出并送入解码器。
- -AugEmbed:移除增强嵌入模块中的转换器,并向互作用模块提供位置和内容嵌入。
- -Interaction:进一步从 AugEmbed 中删除了交互模块。它退化为FCS指纹顶部的解码器。请注意,单独移除交互模块不是有效的模型设计。
- Small:使用较小的数据集来训练FCS:DrugBank用于药物,BindingDB用于蛋白质。我们调整最小频率,以输出与FCS大的相似数量的子结构。
- -FCS:用药物的 ECFP4 和蛋白质的 PSC 描述符替换FCS嵌入。其余模型保持不变,即它们随后被馈入变压器、交互模块和解码器。
我们可以看到CNN、transformer 和交互模块对模型最终性能的贡献。仅FCS指纹就具有很强的交互预测性能。此外,从Small中,我们看到大量未标记的数据是有用的,因为它丰富了输入并提高了性能。从-FCS中,我们看到我们的模型能够适应其他具有类似强大性能的流行指纹。

















 227
227

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









