







小罗碎碎念
今天分享的这篇文章发表在《Nature Cancer》,目前IF=23.5,介绍了一个名为ENLIGHT-DeepPT的深度学习框架,该框架通过模拟转录组学来预测癌症治疗反应。

开头没看懂没有关系,我讲点你能看懂的。你可以粗略的理解为,这篇文章做了一个加法,把两篇文章拼起来了。
上面的这篇文章发表于23年,IF=12.8,利用配对的转录组数据提出了一个无监督学习方法,用于预测患者对特定药物的反应;下面的这个,其实不是文章,而是这篇文献开源的代码,作用是可以不用前期准备配对的数据,直接从HE切片中预测传统借助转录组数据才能预测的癌症治疗反应,从而省去了配对数据的烦恼,如果这个模型真实可行,那么后期研究可以利用的数据将会呈现指数级增长。
最后再解释一下为了我要把第二个模型也视为一篇文章——道理很简单——能实现这个功能的模型不止一个,你完全可以去找其他的文献,来做替换。病理AI目前面临的数据问题其实很多,跨域适配是目前的研究热点,免疫组化+HE,CT+HE,以及MRI+HE,这些不都可以找对应的文献去参照这种模式实现?
为了方便大家积累数据和代码,我直接把开源的数据和代码放在推文的最前面,免得大家翻到最后才能找到。
数据开源
本研究中使用的TCGA组织学图像及其相应的基因表达谱从Genomic Data Commons数据门户(https://portal.gdc.cancer.gov)下载。
TransNEO-breast数据集可在https://ega-archive.org/studies/EGAS00001004582获取。
Trastuzumab1队列是TransNEO-breast数据集的一个子集。Trastuzumab2队列数据从癌症成像档案数据库(https://www.cancerimagingarchive.net)下载。
所有DeepPT预测的表达和相关反应数据,以及计算性能指标的代码,均可在GitHub上获取,网址为https://github.com/PangeaResearch/enlight-deeppt-data。
代码开源
DeepPT框架可在Zenodo上用于学术研究目的,网址为https://doi.org/10.5281/zenodo.11125591(参考文献61)。
ENLIGHT分数可通过网络服务计算,该服务根据肿瘤直接测量或从切片预测的表达谱进行计算,网址为https://ems.pangeabiomed.com/。
文献概述
研究团队由Danh-Tai Hoang、Gal Dinstag、Eldad D. Shulman等人领衔,他们提出了一个两步间接方法:
- DeepPT:一个深度学习模型,可以从H&E染色的肿瘤玻片预测全基因组肿瘤mRNA表达。
- ENLIGHT:利用DeepPT预测出的表达值来预测对靶向和免疫疗法的反应。
研究显示,DeepPT在所有16个癌症基因组图谱(The Cancer Genome Atlas, TCGA)的队列测试中成功预测了转录组学,并在两个独立数据集上表现良好。
ENLIGHT-DeepPT在涉及四种不同治疗方法的五个独立患者队列中成功预测了真正的有反应者,这些队列涵盖了六种癌症类型。总体上,与基线反应率相比,预测反应者的反应率提高了39.5%,整体优势比(odds ratio, OR)为2.28。
文章还讨论了DeepPT预测的基因表达与肿瘤浸润性淋巴细胞(TILs)的相关性,以及如何通过预测的基因表达来推断与患者生存相关的预后特征水平。此外,研究还比较了ENLIGHT-DeepPT的预测准确性与直接从图像预测反应的监督学习方法,并发现两者相当,甚至在某些情况下ENLIGHT-DeepPT表现更好。
这项研究的优势在于它不需要治疗数据就能训练出预测模型,这与通常需要大量匹配的成像和反应数据的监督学习方法形成对比。研究结果表明,结合DeepPT和ENLIGHT的方法为从H&E染色玻片直接预测临床治疗反应提供了一个有前景的新途径。
一、引言
病理学长期以来被认为是癌症临床诊断和预后的金标准。
近年来,肿瘤分子特征的运用使得癌症诊断更加精确,也为精准肿瘤学提供了支持1–3。数字病理学的快速发展,通过应用机器学习和人工智能方法,特别是深度学习在图像分析方面的进展4,使得可以从肿瘤切片中提取临床相关信息。
关键进展已经展开,例如使用苏木精-伊红(H&E)染色的组织全切片图像(WSIs)进行:
- 计算机辅助肿瘤诊断5–8
- 癌症类型分类7,9–13
- 区分低或高突变负荷的肿瘤14
- 识别遗传突变6,15–23
- 预测患者生存率24–30
- 检测DNA甲基化模式31和有丝分裂32
- 量化肿瘤免疫浸润33、肿瘤浸润淋巴细胞(TILs)34和空间免疫细胞浸润35
先前的研究已经令人印象深刻地揭示了利用下一代数字病理学预测治疗反应的潜力36–40。
在这些直接监督学习方法中,从WSI直接预测治疗反应需要包含匹配的成像和反应数据的大型数据集。因此,对于每种要预测的药物或治疗,都需要特定的队列。然而,这类数据的可获得性在大规模上仍然相当有限,限制了这种方法的应用性,并引发了对监督预测器在其他队列中泛化能力的担忧。
为克服这一挑战,作者转向开发并研究一种通用方法,用于生成基于WSI的患者反应预测器,适用于广泛的癌症类型和治疗方法,无需训练时匹配WSI和反应数据集。
为此,作者采取了一种间接的两步法。首先,作者开发了DeepPT(Deep Pathology for Transcriptomics),这是一个深度学习框架,用于从H&E切片预测(估算)基因表达,该框架在先前研究的基础上进行了扩展41–46。DeepPT模型针对特定癌症类型构建,通过训练来自癌症基因组图谱(TCGA)的匹配WSI和表达数据。
其次,对于新患者的基因表达值,作者应用作者先前发表的ENLIGHT方法47,该方法最初用于从测量的肿瘤转录组学预测患者反应,现在用于从DeepPT估算的转录组学预测反应。
接下来,作者概述了DeepPT的架构和ENLIGHT的工作原理,以及研究设计和分析的队列。
DeepPT 架构的三个主要组成部分以及 ENLIGHT 流程的概述。
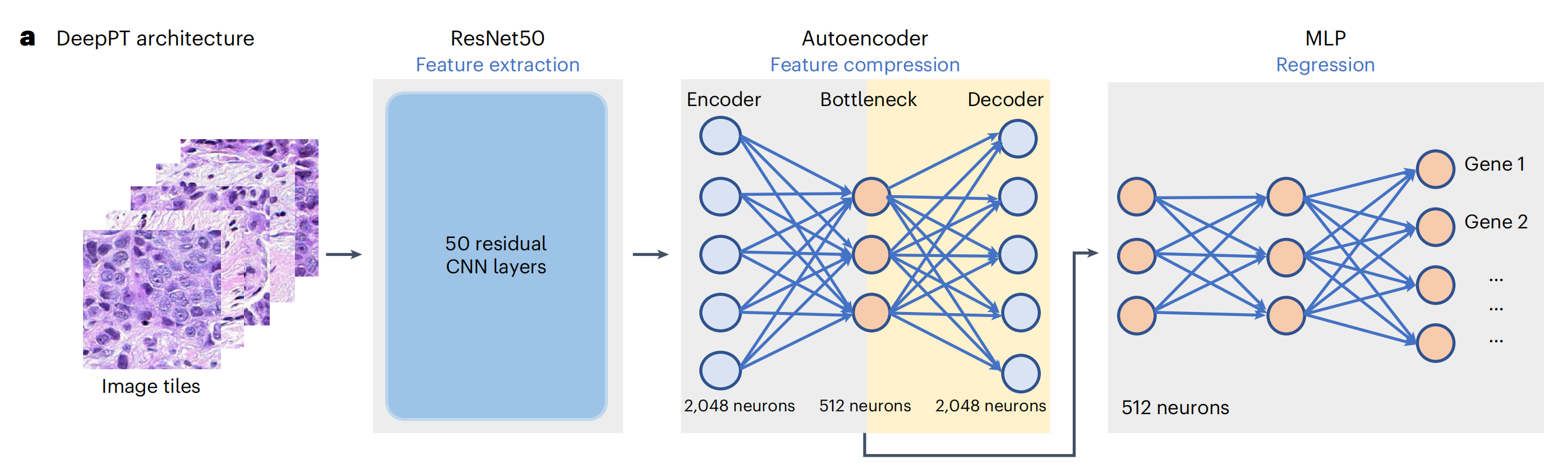
a. DeepPT 架构的三个主要组成部分:
- 预训练的ResNet50卷积神经网络(CNN)单元:这个单元位于左侧,负责从图像瓦片中提取组织病理学特征。ResNet50 是一个深度残差网络,已经在大规模的自然图像数据集(如ImageNet)上进行了预训练,能够识别和提取图像中的复杂特征。
- 自编码器:位于中间,它将 ResNet50 提取的 2,048 个特征压缩到更低维度的 512 个特征。这一步骤有助于去除噪声和冗余信息,同时减少模型过拟合的风险,并降低计算复杂度。
- 多层感知器(MLP):位于右侧,它将压缩后的特征整合起来,预测样本的基因表达。MLP 是一个前馈神经网络,用于学习输入特征和基因表达之间的映射关系。
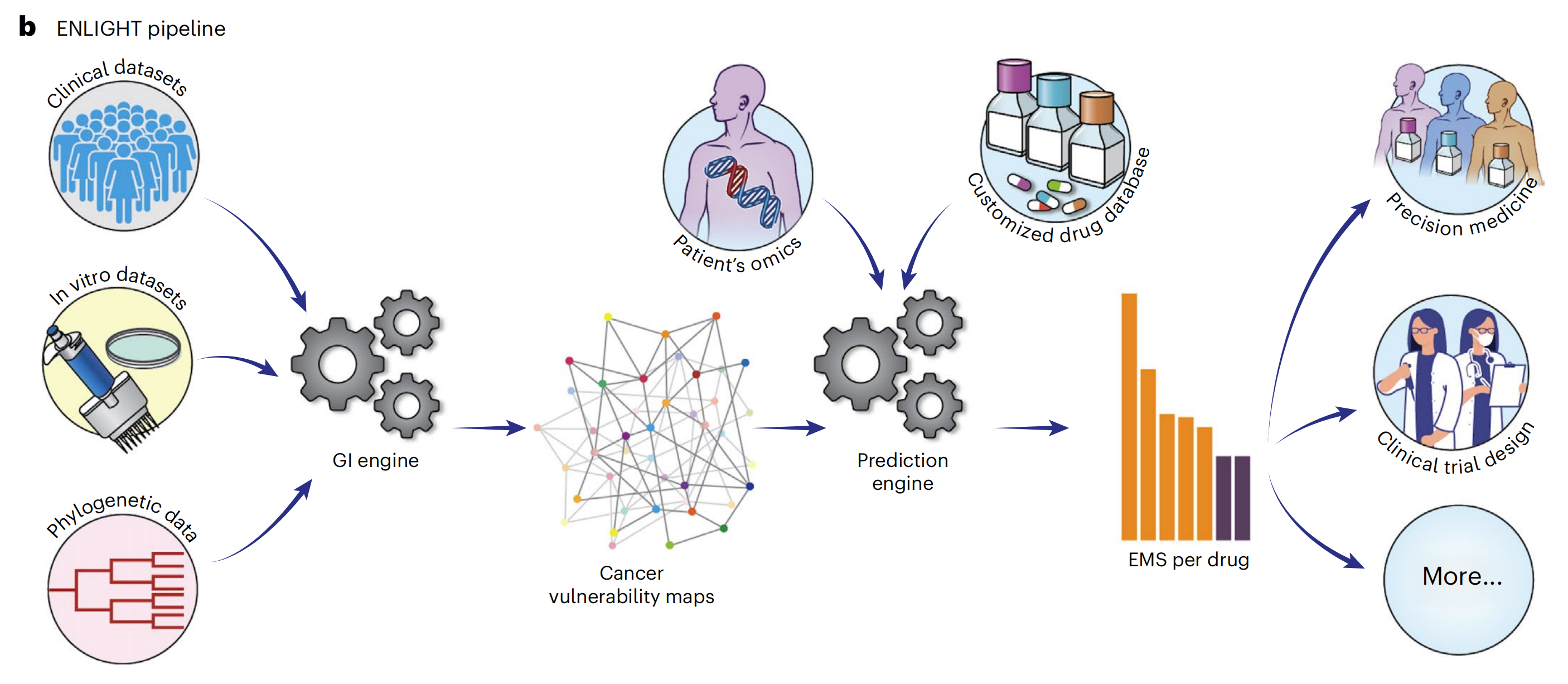
b. ENLIGHT 流程的概述:
- ENLIGHT 是一个无监督学习方法,用于预测患者对特定药物的反应。它首先通过分析体外和临床数据来推断给定药物的目标基因的基因交互(GI)伙伴。
- 给定药物的合成致死(SL)/合成救援(SR)伙伴以及患者样本的转录组学数据,ENLIGHT 计算药物匹配分数,该分数用于预测患者对治疗的反应。
- 在这项研究中,ENLIGHT 使用 DeepPT 预测的表达数据来为每个研究的患者生成药物匹配分数。
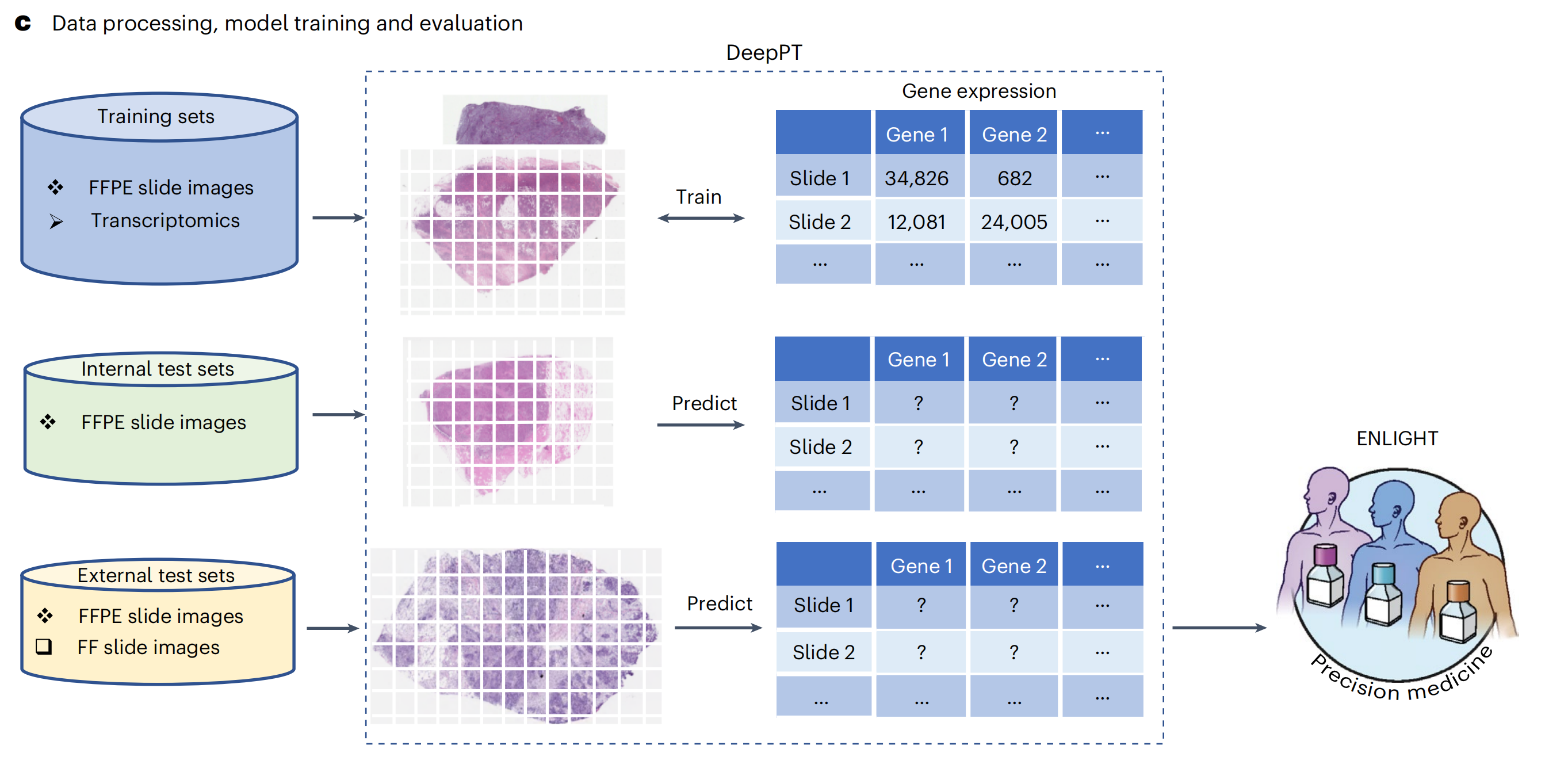
c. 使用 DeepPT 和 ENLIGHT 的分析概述:
- 顶部行:DeepPT 使用来自 TCGA 的多种不同癌症类型的 FFPE 玻片图像和匹配的转录组学数据进行训练。
- 中间行:训练阶段之后,模型被应用于预测内部(保留的)TCGA 数据集以及两个未训练的外部数据集上的基因表达。
- 底部行:在五个独立的测试临床数据集中,预测的肿瘤转录组学作为 ENLIGHT 的输入,用于预测患者对治疗的反应并评估整体预测准确性。
总的来说,Fig. 1 展示了如何通过深度学习模型 DeepPT 从组织学图像预测基因表达,并结合 ENLIGHT 方法来预测癌症患者对特定治疗的反应。这一流程为精准医疗提供了一种潜在的新工具,可以加速从病理图像到治疗决策的过程。
然后,作者描述了ENLIGHT–DeepPT两个步骤中获得的结果。
首先,作者研究了预测肿瘤表达的能力,展示了训练后的DeepPT模型在预测16个TCGA队列和2个独立、未见队列的批量基因表达方面的性能。
其次,作者分析了五个独立的临床试验数据集,这些数据集包含了接受各种靶向和免疫治疗的不同癌症类型的患者。
作者展示了ENLIGHT,在不进行任何调整的情况下,遵循其原始出版物47中的参数,可以成功地基于DeepPT估算的表达值预测真正的应答者。
二、讨论
本研究表明,将DeepPT——一种从H&E切片预测基因表达的深度学习框架,与ENLIGHT——一种已发表的、用于预测预处理肿瘤转录组学生物学响应的无监督计算方法相结合,可以形成一种ENLIGHT–DeepPT方法,用于基于H&E切片预测针对多种靶向和免疫治疗方案的的临床响应。
作者首先展示了DeepPT在从H&E切片预测mRNA表达谱方面显著优于当前最先进的方法。接着,作者展示了ENLIGHT–DeepPT能够成功地从不同适应症的临床数据集中,直接从H&E图像预测真正的应答者,这些数据集中的患者接受了各种靶向药物的治疗。
将DeepPT与ENLIGHT结合用于从H&E切片直接预测响应是一种有前景的方法,因为它不需要在响应数据上进行训练。这与更常见的做法——使用响应数据以端到端的方式训练分类器——相比,是一个关键优势。实际上,将ENLIGHT应用于预测的表达数据已成功使作者能够在跨越六种癌症类型的五个数据集中预测对四种不同治疗的响应。
其次,尽管DeepPT是使用FFPE切片进行训练的,但其泛化能力良好,并且可以不经修改直接用于从FF切片预测表达值。
这证明了DeepPT在预测FF或FFPE切片的RNA表达方面的适用性。然而,尽管这里展示的结果很有希望,但当然,它们应当通过将这里提出的通用流程应用于更多癌症类型和治疗进行进一步测试和扩展。
从H&E切片开发一个响应预测流程,如果具有合理的准确性,并在临床设置中进一步仔细测试和验证,可能会带来极大益处,因为下一代测序通常需要几周时间才能返回结果。
许多晚期癌症患者需要立即治疗,而这种方法有可能在更短的时间内提供治疗选项。然而,应当谨慎注意的是,尽管结果充满希望,但本研究呈现的结果在应用于临床之前,还需在精心设计的前瞻性研究中进行进一步的测试和验证。
作者希望这里呈现的结果既能加速未来的此类努力,也能为使更多此类队列公开可用提供进一步的推动力,以促进此类通用预测方法的进一步发展。
三、方法
3-1:数据收集
本研究使用的数据集来源于公开可获取的资源及内部资源,具体细节如下:
TCGA
TCGA组织学图像及其相应的基因表达谱从Genomic Data Commons数据门户(https://portal.gdc.cancer.gov)下载。仅选择了原发性肿瘤的诊断切片,共计6,269张FFPE切片,涉及5,528名患者,包括:
- 乳腺癌(1,106张切片,1,043名患者)
- 肺癌(1,018张切片,927名患者)
- 脑癌(1,015张切片,574名患者)
- 肾癌(859张切片,836名患者)
- 结直肠癌(514张切片,510名患者)
- 前列腺癌(438张切片,392名患者)
- 胃癌(433张切片,410名患者)
- 头颈癌(430张切片,409名患者)
- 宫颈癌(261张切片,252名患者)
- 胰腺癌(195张切片,175名患者)。
TransNEO-breast
TransNEO-breast数据集包含160名乳腺癌患者的FF切片及其相应的基因表达谱。RNA文库制备和测序协议以及切片数字化的详细信息已在前文描述48。
NCI-brain
NCI-brain组织学图像及其相应的基因表达谱来自NCI病理实验室的档案。这些数据包括226个病例,涵盖各种中枢神经系统肿瘤,包括常见和罕见肿瘤类型。
所有病例均进行了甲基化分析(以评估诊断)和RNA测序。
bintrafusp alfa
bintrafusp alfa治疗队列包括58名患者,其中肺癌(9名患者)、宫颈癌(16名患者)和头颈癌(33名患者)。FFPE切片由NCI提供。
Trastuzumab1
Trastuzumab1队列是上述TransNEO-breast数据集的一个子集,包括64名接受化疗和曲妥珠单抗联合治疗的患者。
Trastuzumab2
Trastuzumab2队列是一个HER2+乳腺癌队列,接受了曲妥珠单抗和化疗的联合治疗,包括85名患者及其FFPE切片40,56。
FFPE切片从癌症成像档案数据库(https://www.cancerimagingarchive.net)下载。
ALKi
ALKi数据集包括14名ALK突变非小细胞肺癌患者,接受了克唑替尼或阿来替尼治疗。相应的FFPE切片由科罗拉多大学提供。
PARPi
PARPi数据集包括13名携带BRCA基因突变胰腺癌的遗传病患者,接受了PARPis治疗。FFPE切片由谢巴医疗中心提供。
每个数据集中,将患者分类为应答者和非应答者是基于运行相应试验的临床医生使用的标准:
- 对于trastuzumab1和trastuzumab2,应答定义为病理完全缓解,而非应答者定义为有残留疾病。
- 对于bintrafusp alfa,应答者定义为部分缓解或完全缓解的患者。
- 对于ALKi,应答定义为无进展生存期超过18个月。
- 对于PARPi,应答定义为总生存期超过36个月。
详细信息可在补充表2中找到。
3-2:组织学图像处理
作者首先使用Sobel边缘检测58来识别每个切片中包含组织的区域。
由于WSIs过大(每个维度有10,000到100,000像素),无法直接输入深度神经网络,因此作者以20×放大率将WSIs分割成512×512 RGB像素的非重叠瓦片。移除超过一半像素的加权梯度幅度小于某一阈值(根据图像质量不同,阈值在10到20之间变化)的瓦片。
根据切片的大小,TCGA队列中每张切片的瓦片数量从100到8,000不等(扩展数据图5a–e)。相比之下,例如TransNEO切片要小得多,每张切片的瓦片数量在100到1,000之间(扩展数据图5f)。
为了最小化染色变异(异质性和批次效应),作者对选定的瓦片应用了Macenko的颜色标准化方法59。
3-3:基因表达处理
基因表达谱以大约60,483个基因标识符的读数形式获得。使用edgeR识别被认为表达的基因,每种癌症类型大约有18,000个基因。每个基因样本间的中位表达量在10到10,000读数之间变化(扩展数据图6a–f)。
为了缩小基因表达值的范围并减少实验和批次之间文库大小的差异,作者进行了如作者先前工作47中描述的标准化处理。
3-4:DeepPT架构
作者的模型架构由三个主要单元组成(扩展数据图1a,b)。
特征提取
作者使用了在ImageNet数据库的1400万自然图像上预训练的ResNet50卷积神经网络模型60来从图像瓦片中提取特征。
在将这些瓦片输入ResNet50单元之前,图像瓦片被调整到224×224像素,以匹配卷积神经网络的标准输入尺寸。
通过特征提取过程,每个输入瓦片由一个包含2048个衍生特征的向量表示。ResNet50预训练模型在包含约1400万张图像的ImageNet等大型数据集上进行了训练,跨越1000个标签。它作为实现迁移学习的基本元素,在数字病理学中得到了广泛应用。
在DeepPT的背景下,作者去掉了ResNet50的最后几层,以确保提取的特征更具通用性,更适合数字病理学任务,而不是受限于原始的ImageNet分类任务。
特征压缩
作者应用了一个包含512个神经元的瓶颈层的自编码器,将特征数量从2048减少到512。这有助于排除噪声,避免过拟合,并减少计算需求。
如扩展数据图7a,b所示,大量的ResNet特征经常为零。这种数据稀疏性在自编码器特征中显著减少(扩展数据图7c,d)。
为了证明自编码器模块的优势,作者进行了一项实验,从作者的框架中排除自编码器模块,在TCGA-breast队列上训练模型,同时保留ResNet50特征,并在TransNEO-breast队列上测试它。这个模型的预测准确率(ResNet)明显低于保留了自编码器组件的模型,如DeepPT中所做的那样(ResNet+自编码器)(扩展数据图7e)。
这一趋势在NCI-brain队列中也一致观察到,强调了自编码器模块的益处。
多层感知器回归
此组件的目的是构建一个预测模型,将上述自编码特征与全基因组基因表达联系起来。
该模型包括三个层次:
- (1) 一个输入层,包含512个节点,反映自编码向量的尺寸;
- (2) 一个隐藏层,其大小取决于共同考虑的基因数量;
- (3) 一个输出层,每个基因对应一个节点。
由于训练数据包含切片级别的基因表达(即批量基因表达,而不是空间分辨率),作者平均了每个瓦片的预测,以获得切片级别的平均值。
3-5:DeepPT训练和评估
作者独立地针对每种癌症类型进行训练和评估。
为了评估作者模型的性能,作者应用了5×5嵌套交叉验证。对于每个外循环,作者将整个患者群体(每个队列)划分为训练(80%)和保留测试(20%)集。
作者进一步将训练集划分为内部训练和评估集,按照五折交叉验证进行。每个训练/验证集对模型进行独立训练和评估。五个不同模型的预测平均值代表作者对每个保留测试集中的每个单个基因的最终预测。作者对五个保留测试集重复此过程,共训练了25个模型。
这些在TCGA队列上训练的模型用于预测给定外部队列中每个基因的表达,通过计算所有模型预测值的平均值来实现(扩展数据图1c,d和8)。
由于每个患者可以有多张切片,作者将切片级别的预测平均以获得患者级别的预测。
每个训练轮次在达到500个epochs之前停止,或者如果验证集上实际与预测的基因表达值之间的平均相关性在连续50个epochs内没有改善,则更早停止。
在自编码器和多层感知器(MLP)回归模型中使用了Adam优化器和均方误差损失函数。自编码器模型和MLP回归模型的学习率均为0.0001,每个步骤使用32个图像瓦片作为minibatch大小。
为了减少过拟合,作者使用了0.2的dropout。
3-6:数据增强
由于TCGA-胰腺腺癌队列的样本数量相对较少,为了进一步减少过拟合,作者通过将WSIs旋转90°、180°和270°来人为增加这个队列的数据量。
在测试时间,四个对称的平均值代表作者对每个切片的预测。
由于计算需求高,其他队列未进行数据增强。
3-7:直接监督模型
直接(端到端)监督模型旨在直接从切片中分类应答者和非应答者,而无需进行基因表达预测的中间步骤。为此,作者应用了用于预测基因表达的相同计算深度学习框架,只是将MLP回归组件替换为MLP分类组件。
每个评估数据集都独立处理。根据先前的方法6,9,20,23,45,所有瓦片继承了其所属切片的标签。由于每个数据集的样本数量相对较少,作者使用留一交叉验证来评估直接监督模型。
对于每个保留的患者,作者使用bootstrap采样技术将剩余的患者随机划分为训练(80%)和验证(20%)集30次,从而得到30个模型。
对于每个模型,切片区预测是通过该切片内所有瓦片级别的预测的平均值计算得到的。每个保留切片的最终预测是通过在30个模型上计算其预测值的平均值得到的。
3-8:实现细节
本研究中所有分析均使用Python 3.9.7和R 4.1.0进行,包括Numpy 1.20.3、Pandas 1.3.4、Scikit-learn 1.1.1、Matplotlib 3.4.3和edgeR 3.28.0。
图像处理,包括瓦片分割和颜色标准化,使用OpenSlide 1.1.2、OpenCV 4.5.4和PIL 8.4.0。
特征提取、特征压缩(自编码器单元)和MLP回归部分使用PyTorch 1.12.0实现。
Pearson相关性使用Scipy 1.5.0计算。
3-9:ENLIGHT
ENLIGHT的药物响应预测包括两个步骤。
(1)给定一种药物,GI引擎识别药物靶基因的临床相关GI伙伴。
GI引擎首先通过分析癌症细胞系依赖性,根据SL/SR相互作用应分别降低/增加肿瘤细胞活力的原则,识别出一系列初始候选合成致死(SL)/合成拯救(SR)相互作用。
然后,通过分析与转录组学和生存数据相关的肿瘤样本数据库,选择那些更可能具有临床相关性的对,需要目标基因和伙伴基因的共同失活与更好的患者生存之间存在显著关联,对于SL相互作用和类似地对于SR相互作用。
(2)然后使用药物特定的GI伙伴来预测患者对每种药物的响应,基于患者肿瘤的基因表达谱。
EMS,用于评估患者与治疗的匹配度,是基于从基因表达推断出的药物靶基因GI伙伴基因集的整体激活状态,反映了这样一个理念:肿瘤对诱导更多活跃的SL相互作用和更少活跃的SR相互作用的药物更为敏感。
3-10:统计学和可重复性
在TCGA中,由于H&E切片的质量较低,从每个队列的分析中排除了从每个队列的分析中排除了7至100名患者。在其他情况下,没有数据从分析中排除。
没有使用统计方法预先确定样本大小,但作者的样本大小与以前发表的论文44,49中的样本大小相似。文章中指定了使用的统计测试和方法以及样本大小。所有数据均符合所使用的统计测试的假设。
在整个文章中,P值都进行了假发现率(FDR)校正,如个别指定的。路径富集分析、生存分析、OR和AP的统计方法详细如下。数据收集和分析未对实验条件进行盲法。
3-11:路径富集分析
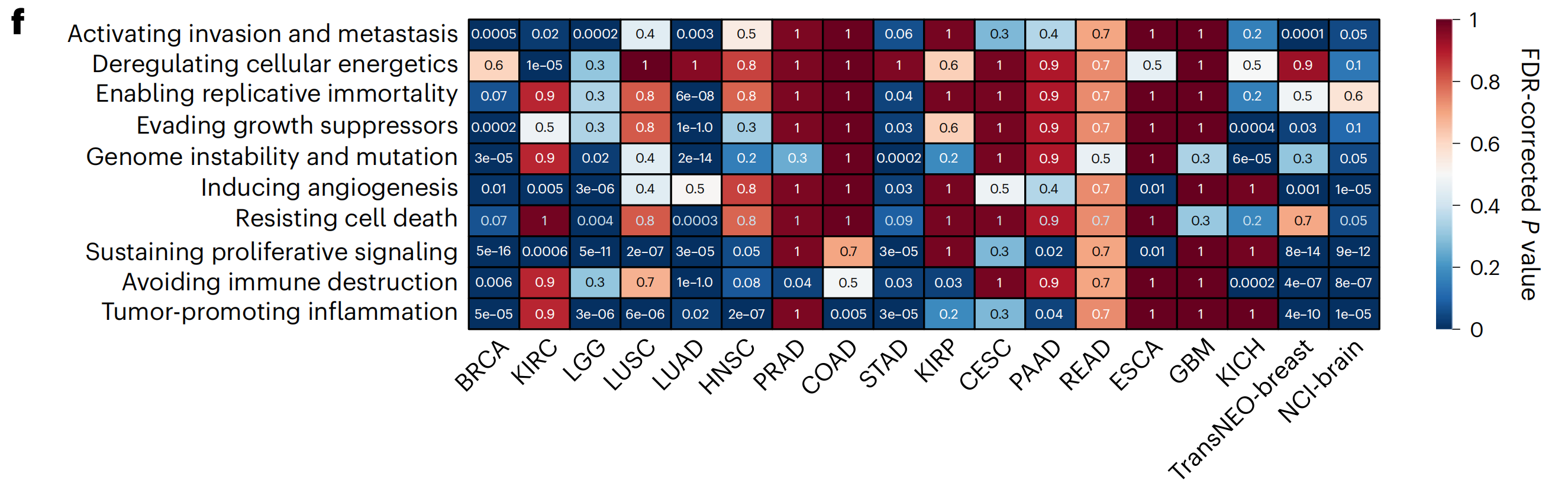
为了测试DeepPT高度预测的基因(R>0.4)是否富集于癌症的特征,作者进行了十种癌症特征的hypergeometric测试,这些特征在参考文献50中描述,并在参考文献51中给出了详细的基因集。
然后,作者使用Benjamini-Hochberg FDR对P值进行了校正。结果在图2f中展示。
3-12:生存分析
作者使用Cox比例风险模型计算了基于实际和预测表达的三个预后标志的HRs,如在图4中展示。
Fig. 4 展示了基于实际和预测表达的三种增殖特征(Ki-67、增殖指数和EMT途径)的生存相关性相关性比较。
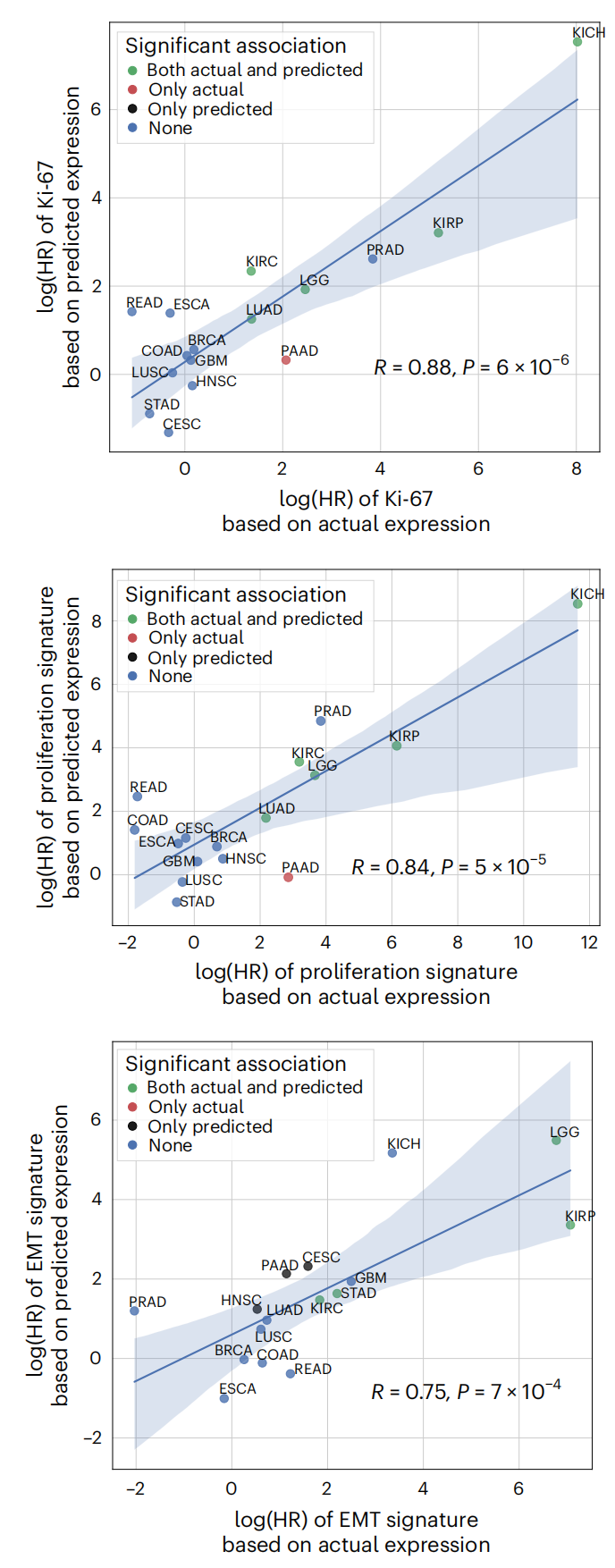
以下是对图的详细分析:
-
X轴和Y轴:
- X轴表示基于实际表达的签名得分的对数风险比(log(HR))。
- Y轴表示基于预测表达的签名得分的对数风险比。
-
数据点:
- 每个数据点代表不同的癌症基因组图谱(TCGA)队列。
- 数据点根据生存关联的显著性被着色编码:
- 绿色:实际和预测签名的生存关联都显著。
- 红色:仅实际签名的生存关联显著。
- 黑色:仅预测签名的生存关联显著。
-
显著性阈值:
- 使用校正后的 P 值小于 0.05 作为显著性阈值。
-
相关性:
- 每个面板展示了实际表达和预测表达之间的皮尔逊相关系数(R)和相应的 P 值,这些值衡量了两种表达方式之间生存风险比的一致性。
-
回归线和置信区间:
- 显示了数据点的线性回归线以及 95% 置信区间,这有助于评估预测表达的生存关联与实际表达的生存关联的接近程度。
-
三个增殖特征:
- 顶部(Ki-67):Ki-67 是一个已知的细胞增殖标记物,广泛用于多种癌症类型中,包括乳腺癌、结直肠癌、肉瘤、鳞状细胞癌和前列腺癌。
- 中间(增殖指数):增殖指数是从文献中推导出的,用于衡量细胞增殖水平,并且在多种癌症中被证明具有预后价值。
- 底部(EMT途径):上皮-间质转化(EMT)途径与肿瘤的侵袭和转移有关,也在多种癌症中显示出预后相关性。
Fig. 4 的关键信息是 DeepPT 模型不仅能够预测基因表达,而且预测的表达能够与实际表达一样,保持与患者生存相关的预后价值。
这表明 DeepPT 模型在预测癌症治疗反应方面的潜力,并且其预测结果可以作为实际生物标志物的可靠替代。
3-13:OR
在临床环境中,OR是两个不相交组对治疗响应的比率的比率。具体来说,给定:
A. ENLIGHT匹配患者中治疗响应的概率
B. ENLIGHT匹配患者中无响应的概率
C. ENLIGHT不匹配患者中治疗响应的概率
D. ENLIGHT不匹配患者中无响应的概率
OR是(A/B)/(C/D)。


















 1373
1373

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









