







目录
4.2扩张的卷积神经网络(Dilated convolutional neural network)
4.4Site prediction based on dilated convolutional Bidirectional LSTM
5.1Comparison with other different learning models
EMDLP: Ensemble multiscale deep learning model for RNA methylation site prediction
期刊:BMC Bioinformatics
中科院分区:3Q
影响因子:3.307
web网页:http://www.labiip.net/EMDLP/index.php (http://47. 104.130.81/EMDLP/index.php)
Github:无
1.摘要
背景:最近的研究建议,通过转录后RNA修饰进行表演转录组调节对于各种RNA至关重要。 RNA修饰的确切识别对于理解其目的和调节机制至关重要。但是,鉴定RNA修饰位点的传统实验方法相对复杂,耗时且费力。机器学习方法已应用于RNA序列的过程中,以计算方式提取和分类,这可以更有效地补充实验方法。最近,由于其在表示学习中的强大功能,已证明了卷积神经网络(CNN)和长期短期记忆(LSTM)在修改站点预测中的成就。但是,CNN可以从空间数据中学习局部响应,但不能学习顺序相关。 LSTM专门用于顺序建模,并且可以访问上下文表示,但与CNN相比缺乏空间数据提取。出于这些原因,使用自然语言处理(NLP),深度学习(DL)构建预测框架有很大的动力。结果:本研究提出了一个集合多尺度深度学习预测指标(EMDLP),以NLP和DL方式识别RNA甲基化位点。它有机地结合了扩张的卷积和双向LSTM(BILSTM),这有助于更好地利用本地和全球信息进行现场预测。 EMDLP的第一步是以NLP方式表示RNA序列。因此,从本地和全局信息的角度来采用了三个编码,例如RNA单词嵌入,单热编码和RGLOVE,这是基于手套的单词向量表示的一种改进的学习方法。然后,使用扩张的卷积神经网络(DCNN)构建扩张的卷积双向LSTM网络(DCB)模型,然后是Bilstm,以提取甲基化位点预测的潜在特征。最后,这三种编码方法是通过软投票集成的,以获得更好的预测性能。 M1A和M6A的实验结果表明,EMDLP的接收器操作特征(AUROC)下的面积分别获得95.56%,85.24%,并且表现优于最先进的模型。为了最大化用户的便利性,EMDLP的用户友好网络服务器已公开不可用。
2.背景
RNA分子的功能多样性通过转录后RNA的修饰比较丰富,从而调节RNA寿命的所有阶段[1]。到目前为止,已经发现了大约160种不同形式的RNA修饰[2],包括N1-甲基二苯胺(M1A),N6-甲基腺苷(M6A),5-甲基辛托斯(M5C),N2-甲基糖苷(N2-甲基糖苷) 7-甲基鸟苷(M7G)[3,4]等。其中,M1A修饰是一种普遍的RNA修饰,发生在与甲基连接的腺嘌呤碱基的氮-1位置上发生[5],如图图所示。1A。它与呼吸链,神经发育回归和介导抗生素耐药性细菌等问题有关[6-8]。影响腺嘌呤的另一种修饰是M6A修饰,这是哺乳动物中最丰富的修饰,它发生在腺苷碱的氮-6位置上[9],如图1B所示。它对人类的生长和疾病有深远的影响[10]。腺苷通常经历M1A和M6A [11]。有趣的是,在碱性条件下,也已知M1A会经历Dimroth重排向M6A [11]。因此,重要的是要准确识别M1A和M6A修饰位点,以发现这些修饰的机制和功能[12]。
已经构建了许多用于识别M1A和M6A修饰位点的实验方法,并在高通量测序技术方面取得了重大进展,例如M6A-CLIP [13],M6A-MICLIP [14],M1A-SEQ [15],M1A-ID -seq [11]等。但是,实验方法昂贵且耗时,这限制了它们的广泛使用[16]。幸运的是,各种计算方法已成为该领域的强大补充。
大多数为从序列中预测位点而设计的机器学习方法通常首先基于人类理解的特征方法提取特征,然后是分类器来预测该位点是否是甲基化位点。例如,RAMPred基于核苷酸化学性质(NCP)、核苷酸组成(NC)提取特征,并首次采用支持向量机(SVM)预测m1A甲基化位点[17]。iRNA-3typeA基于NCP、累积核苷酸频率(ANF)提取特征,并采用SVM预测m1A、m6A和A-to-I修饰位点[18]。iMRM基于NCP、NC、One-hot编码、二核苷酸二进制编码(DBE)、核苷酸密度(ND)、二核苷酸理化性质(DPCP)提取特征,采用极端梯度增强(XGboost)预测m1A、m6A、m5C、ψ和A-to-I修饰位点,性能优于现有方法。
M6AMRF基于DBE提取的特征,使用了F-评分算法与顺序前向搜索(SFS)相结合来提高特征表示,并使用XGBoost来预测M6A位点[20]。 Rnamethpre首先提取了侧翼序列,局部二级结构数据和相对位置数据的特征,然后采用SVM来预测具有令人满意的性能的M6A甲基化位点[21]。 SRAMP通过利用单速编码,k-neart邻居编码和核苷酸对频谱编码来预测M6A位点来结合三个随机森林分类器[22]。 RFATHM6A根据四种编码方法提取的特征,包括Knucleotide频率(KNF),位置特异性核苷酸序列曲线(PSNSP),Kspaced核苷酸对频率(KSNPF)和位置特异性的Dinucleotide序列(PSDSP),然后分别构建与Athmethpre,M6ath和Ramnpps相比,随机森林模型具有竞争力[23]。 Whistle除了整合常规序列特征外,还增加了35个基因组特征,并通过SVM预测M6A甲基化[24],与其他计算方法相比,它显着改善。但是,只有仅提供了几个RNA序列来预测M6A甲基化时,基因组特征并不总是可用的。这些结论表明,提取的特征对于最终预测至关重要。
众所周知,RNA-Seq包含丰富的生物特征信息。因此,RNA序列的理性表示变得更加关键。为了解决这个问题,通过自然语言处理(NLP)对序列的表示形式吸引了很多关注[25],其中RNA序列被视为句子,并且K- Monoricer单元(K-MER)被视为一个词,获得了很大的吸引力[26,27]。与传统的机器学习方法相比,大多数深度学习(DL)模型可以分为三个部分:首先,通过NLP模型学习输入数据表示[28];其次,撰写已经学到的矢量[29];第三,分类器分类以预测该位点是否是甲基化位点。
到目前为止,已经开发了一些使用NLP和DL网络的预测方法来预测m6A或m1A位点。其中,Gene2Vec [30]、DeepPromise [12]和EDLm6Apred [16]是最具代表性和先进性的甲基化位点预测方法。具体来说,基于Word2vec [31]和卷积神经网络(CNN)开发了Gene2Vec来预测m6A位点。DeepPromise采用了CNN并集成了增强核酸内容(ENAC) [32]、RNA字嵌入[33]和一键编码[20,34]功能来识别m1A和m6A位点。EDLm6Apred采用了Word2vec,One-hot编码,RNA字嵌入,BiLSTM来预测m6A位点。然而,现有的方法具有以下缺点。众所周知,从自然语言处理的角度来看,ENAC、One-hot和RNA单词嵌入关注的是局部语义信息[16],而忽略了上下文和全局信息。Word2vec编码考虑了上下文窗口信息,忽略了全局信息[35]。从DL的角度来看,CNN可以从空间数据中学习本地响应[25]。卷积核的不同尺度影响网络的学习能力。Gene2Vec [30]和DeepPromise [12]直接使用单尺度卷积核组成的CNN,可能导致序列的不完全表征学习[36]。这两种方法中缺少的信息对最终的场地预测可能是重要的。此外,CNN没有记忆功能,缺乏学习序列相关性的能力[25]。相反,EDLM6APRED [16]提出了一个深层的Bilstm网络,以解决上述问题,同时访问了上下文信息。但是,Bilstm与CNN相比缺乏空间数据提取,并且需要高训练时间。
考虑以上问题。本文提出了EMDLP以NLP和DL方式鉴定RNA甲基化位点。具体而言,最初使用单速编码,RNA单词嵌入和rglove编码序列。其次,用DCNN构建了DCB模型,然后是BilstM,以提取甲基化位点预测的潜在特征。第三,根据DCB模型构建了三个预测因子,该预测因素是通过上面的三个特征编码方法构建的。最后,EMDLP是通过软投票制定的,平均预测概率使用三个预测因子来获得更好的预测性能。结果表明,在独立测试中,EMDLP模型的性能优于诸如Deeppromise [12]和EDLM6APRED [16]之类的最新方法。
3.数据集
我们提取了在单核苷酸分辨率上发表的两种常见的人RNA修饰站点数据集,包括M1A和M6A。对于M1A和M6A位点,本文中的数据集源自Chen等人的先前研究。 [12]和Zou等。 [30]分别。唯一的区别是,Zou验证集用作M6A站点上本文的独立测试集。
该研究将数据集分为两个部分:用于交叉验证测试的基准数据集和用于独立测试的独立数据集。它将修改的/非修饰站点作为每个样品的中心,并带来了(2n + 1)-nt部分序列窗口。值得注意的是,这两个修改的“ n”不同。指的是Chen论文中的实验结果,最佳窗口的大小分别为M1A和M6A位点的101和1001 [12]。如果原始序列的长度短于2n + 1,则空位置将用字符“ - ”填充,以确保序列长度一致。 M1A位点和M6A位点的正和负样品的比率分别为1:10和1:1。表6显示了这两个RNA修改数据集的统计量。

4.方法
4.1特征提取
One-hot和GloVe
One-hot编码是一个稀疏的二进制、高维的词向量,而RNA词嵌入是一个连续的、低维的密集的词向量,它捕获了局部的语义信息。RGloVe继承了GloVe的原理,捕捉全局语义信息。
RNA单词嵌入是编码RNA序列的标准方法。 RNA序列上的大小K幻灯片的滑动窗口通过重叠相等的长度以形成K-MER子序列,并将这些子序列作为词汇创建。以M1A为例。通过大小3的滑动窗口将101nts的序列转换为99个子序列,该研究获得了105个不同的子序列,这些子序列由唯一的整数索引索引。每个预处理的序列都用一个整数索引更改,并将其馈入KERAS嵌入层以生成300维单词向量。因此,将101nts序列转换为99×300的基质。
嵌入RNA单词仅考虑频率信息,但忽略了上下文和全局信息。 Word2Vec仅通过从每个本地上下文窗口中的信息独立训练,而它不使用全局共发生矩阵中的统计数据[35]。 Pennington等。 [40]拟议的全球向量(手套)可以考虑全局共发生矩阵中的统计数据,并使用Adagrad训练手套单词嵌入[41]。但是,Adagrad的主要弱点可能会导致Adagrad的学习率降低并变得非常小,此时算法无法学习新信息[41]。因此,该研究使用RMSPROP而不是Adagrad来最大程度地减少全局矢量模型的损耗函数。通过这种方法训练的矢量一词称为rglove。特定的分析过程如下。
4.2扩张的卷积神经网络(Dilated convolutional neural network)
Holschneider等。 [43]是第一个发展扩张卷积的人,该卷积通过将孔引入常规卷积[44]来保持特征图的分辨率。与普通的卷积相比,扩张的卷积增加了一个名为扩张率(DR)的高参数,该量表与内核间隔的数量相对应,例如普通卷积中的DR = 1。
除了扩张的卷积外,DCNN还包括合并和辍学层。池层应用于每个特征映射,并输出池窗口中输入的平均值或最大值,以便池化层可以减少参数数量。
辍学层用于避免在模型训练期间过度拟合,并且是最常用的正则化技术。在转发传播过程中的每个训练活动中,一些神经元被随机设置为零,这直觉从而导致不同网络的整合。辍学率是神经元退出的概率。在这项研究中,将三个扩张速率的卷积层(分别为DR = 1、2和3)连接到BilstM阶段。
4.3Bidirectional LSTM
Bilstm是一种结合前向LSTM和向后LSTM的特定反复神经网络(RNN)。其中,正向LSTM计算向前方向上的隐藏特征,并在每一刻 - →H2, - →H3,...→H5时保存输出。通过相同的推理,向后LSTM在反向方向上计算了隐藏的特征,并在每时每刻← - H5,←-H4,...←-H2中保存输出,如附加文件1:图S2所示。最终,最终结果是由在每个瞬间合并向前和向后LSTM层的输出值。 LSTM [45]框架解决了RNN中的爆炸或消失梯度。通常,LSTM单元定义为当前输入XT,内存单元,一个输入调制向量̃ ct,隐藏的状态,忘记门ft,输入门口,一个输出门口和输出门t,如图所示附加文件1:图S3。其中,存储器单元CT由三个“门”控制:忘记门ft,输入门和一个输出门户,其中其条目在[0,1]中。以下是LSTM过渡方程:
4.4Site prediction based on dilated convolutional Bidirectional LSTM

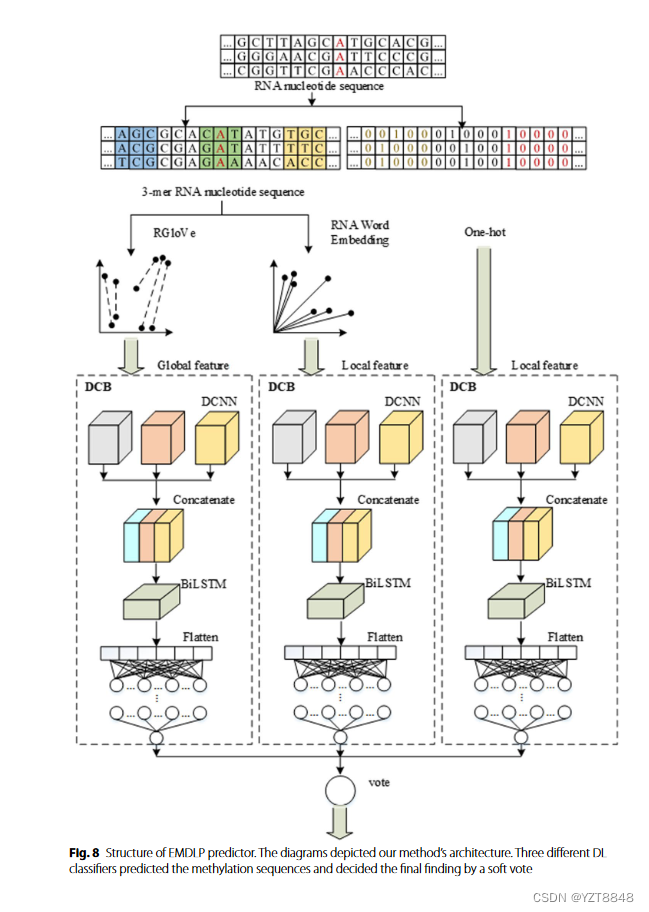
5.结果
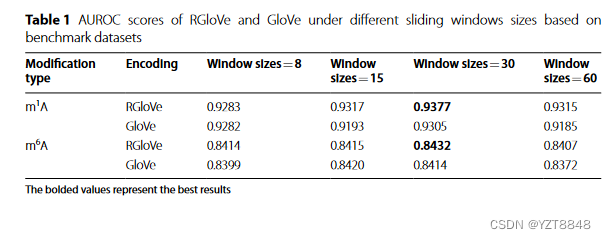
本文首先检查了rglove和手套在不同滑动窗口尺寸上的性能。其次,比较了自制的DCB模型,并与CNN,DCNN和BILSTM模型进行了分析。第三,这项研究将RGLOVE的特征与另外三个预测甲基化修饰位点进行了比较。最后,本文将EMDLP模型与基于独立数据集的最新方法进行了比较。我们的计算设备具有两个NVIDIA RTX2080TI GPU和11 GB的GPU设备内存。除GPU外,该机器还具有两个2.3 GHz 16核Intel(R)Xeon(R)Gold 5218 CPU和128 GB RAM。该设备配备64位Windows10专业版20H2,Python 3.7.6,Keras 2.2.4和Tensorflow-GPU 1.14.0。滑动窗口的大小是影响编码方案性能的重要参数。基于基准数据集,该实验比较了在四个不同的滑动窗口大小下(即8、15、30和60)预测M1A和M6A甲基化位点时的RGlove和Glove的性能。 rglove基于手套模型框架,并采用RMSPROP而不是Adagrad来最大程度地减少全局向量模型的损耗函数。结果,当滑动窗口长度= 30时,rglove显示出最佳的预测性能,如表1所示。实验结果表明,使用RMSPROP可以更有效地训练模型。
5.1Comparison with other different learning models
接下来,使用相同的基准数据集比较DCB并将DCB与CNN,DCNN和BILSTM分析。该实验分别使用rglove编码来描述RNA序列,构建的cnnrglove,dcnn rglove,bilstm rglove和dcbrglove。其中,Cnnrglove采用了CNN模型,用于降低[12]。 dcbrglove代表了一个自建造的DCB模型,包括DCNN和BilstM阶段。 dcnnrglaged表示dcbrglove删除了Bilstm阶段,该阶段被平坦的层代替。同样,Bilstmrglove代表没有DCNN阶段的DCBRGLOVE。图2和表2中显示了M1A和M6A上的AUROC和AUPRC曲线的五倍交叉验证评估。 Dcnnrglove的AUPRC比Cnnrglove高0.08%和0.94%。这个结果。
验证CNN中的单尺度卷积内核是从RNA序列中学习深层语义的挑战。相反,多尺度卷积内核可以提取其他功能以提供深层语义。此外,该研究比较了dcbrglove和dcnnrglove的性能。 DCBRGLOVE的AUROC分别比M1A和M6A上的Dcnnrglove高0.72%和0.77%,而DCBRGLOVE的AUPRC分别为2.01%和0.96%,高于M1A和M6A上的Dcnnrglove。原因可能是DCNN没有内存函数,无法学习顺序相关。相反,DCB可以根据DCNN捕获不同空间结构的局部相关性,并根据Bilstm有效地学习文本中每个K-MER的上下文。总而言之,与其他方法相比,DCB可以更准确地理解序列语义。
最后,研究比较了DCBRGloVe和BiLSTMRGloVe的运行时间。虽然影响模型训练时间的因素很多,但实验结果表明,BiLSTMRGloVe的训练时间很长,是DCBRGloVe的几倍。原因是DCNN级的最大池层减少了网络的参数,对降低维数和计算复杂度起到了积极的作用。

6.discussion
本文提出EMDLP以NLP和DL方式识别RNA甲基化位点。具体讨论如下:首先,本研究比较了基于RGloVe和GloVe编码方法的四种不同滑动窗口大小(即8、15、30和60)下预测m1A和m6A甲基化位点的性能。评估结果表明,使用RMSProp代替Adagrad来最小化全局向量模型的损失函数确实可以更有效地训练模型。这个结果与Ruder,S. (2017)指出RMSProp可以克服Adagrad的弱点是一致的。当滑动窗口长度= 30时,RGloVe显示出最佳的预测性能。
其次,基于上述RGLOVE的序列的特征表示,本研究将DCB模型与CNN,DCNN和BILSTM模型进行了比较,以预测甲基化修饰位点。实验结果表明,dcnnrglove的AUROC比M1A和M6A上的Cnnrglove高0.57%和0.74%。这项研究证实,多尺度卷积内核可以提取不同的特征以提供深层语义。实验结果表明,Bilstmrglove的训练时间很长,并且是DCBRGLOVE的几倍。这也符合最小X.的结论,这表明DCNN阶段的最大流动层减少了网络的参数,该参数在降低维度和计算复杂性方面起着积极作用。实验结果表明,在预测M1A和M6A位点时,DCBRGLOVOVE模型优于其他模型。这项研究证实,DCNN和BilstM的组合使对序列语义的理解更加准确。
第三,基于上述自行构建的DCB模型,本文比较了rglove,RNA单词嵌入,一hot编码和Word2Vec的预测性能。结果表明,我们的RGLOVE在预测性能方面的表现优于其他三个编码方案。这一发现与Pennington,J(2014)一致,后者提出手套的语义精度比Word2Vec更高。最后,EMDLP是通过软投票来构建的,使用三个预测指标获得更好的预测性能。本文根据独立数据集比较了EMDLP,DEEPPROMISE,DCBDEEPPROMISE和EDLM6APRED的预测性能。结果表明,EMDLP的AUROC明显优于三种方法。这项研究进一步表明,与其他四个编码相比,rglove可以更好地表示序列的语义信息,并且与其他方法相比,DCB更适合于提取RNA序列的特征。
7.Conclusions
本文的贡献提出了预测指标EMDLP,以通过NLP和DL方式识别RNA甲基化位点。它有机地结合了扩张的卷积和BilstM,这有助于更好地利用本地和全球信息进行现场预测。尽管EMDLP优于最先进的预测因子,该预测因子目前仅限于人类,并且由于缺乏足够数量的其他物种的单核苷酸数据集,因此尚未扩展到其他模型生物体。值得期待测试EMDLP的性能时,将来有足够的其他物种RNA修饰数据集可用。
没有代码数据集做不了,况且结果也比较高,文章期刊也不是很好。















 1197
1197











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









